Chapter 5 Przykłady doświadczalne
W §5.2 przedstawiono użycie metod ab initio do określania mikrostruktury i jednowymiarowego nieuporządkowania w nanoproszkach , w oparciu o prezentowane w części teoretycznej zmodyfikowane równanie Debye’a oraz algorytmy genetyczne.
W §5.3 wykorzystano metodę do wykonania dużej liczby oznaczeń rozkładu wielkości ziaren nanoproszków i syntezowanych różnymi technikami. Zbadano korelacje pomiędzy metodą otrzymywania materiału a postacią rozkładu wielkości ziaren. Okazuje się, że względna szerokość rozkładu wielkości ziaren może być odciskiem palca metody syntezy nanoproszku.
W §5.4 pokazano, że rozpraszanie niskokątowe jest techniką komplementarną do badania profili refleksów bragowskich. Jest ono źródłem informacji o fraktalnej mikrostrukturze nanoproszków. Ustalono wartość masowego wymiaru fraktalnego oraz zbadano proces degeneracji (łamania) fraktala w czasie wysokociśnieniowego zagęszczania proszków i .
Ostatnim doświadczeniem, opisanym w §5.5, jest wysokociśnieniowa przemiana strukturalna nanokryształów polegająca na tworzeniu się błędów ułożenia. Zaproponowano prosty mechanizm relaksacji naprężeń polegający na losowych przesunięciach warstw kryształu. Symulacja takiego procesu relaksacji połączona z obliczeniami ab initio dyfraktogramów pozwala zrekonstruować obserwowane doświadczalnie linie dyfrakcyjne o asymetrycznych i złożonych profilach, a co za tym idzie - ilościowo opisać przemianę strukturalną.
Zebrany materiał doświadczalny został pomyślany przede wszystkim jako ilustracja podanego w części teoretycznej opisu dyfrakcji proszkowej uwzględniającego “najważniejszy z defektów” kryształu: jego brzeg. Rozdział ten stanowi jednocześnie przegląd informacji o mikrostrukturze nanokrystalicznych proszków , i oraz jej przemian pod wpływem wysokiego ciśnienia.
5.1 Definicje, materiały i metody
5.1.1 Struktura najgęstszego upakowania (close packing)
Struktura atomowa węglika krzemu , azotku galu i jest strukturą najgęstszego upakowania, podobnie jak w modelowej strukturze blendy cynkowej (), rys. 5.1. Sieć krystaliczna jest zbudowana z dwóch identycznych podsieci cynku i siarki. W węgliku krzemu są to odpowiednio podsieci węgla i krzemu. W diamencie obie podsieci są obsadzone atomami węgla.
a)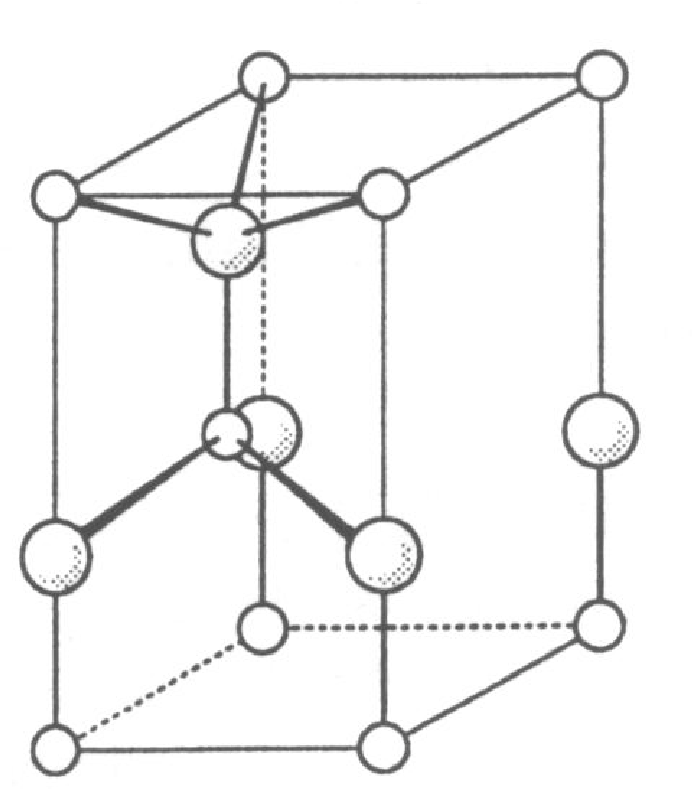
|
b)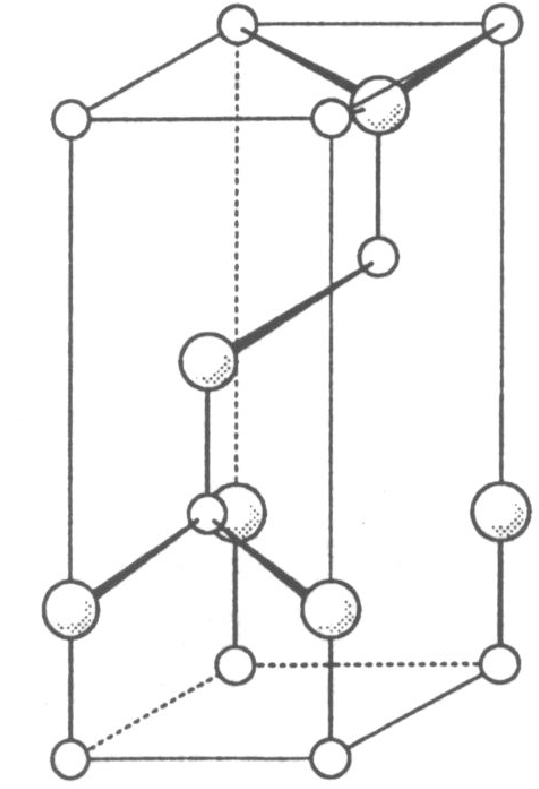
|
c)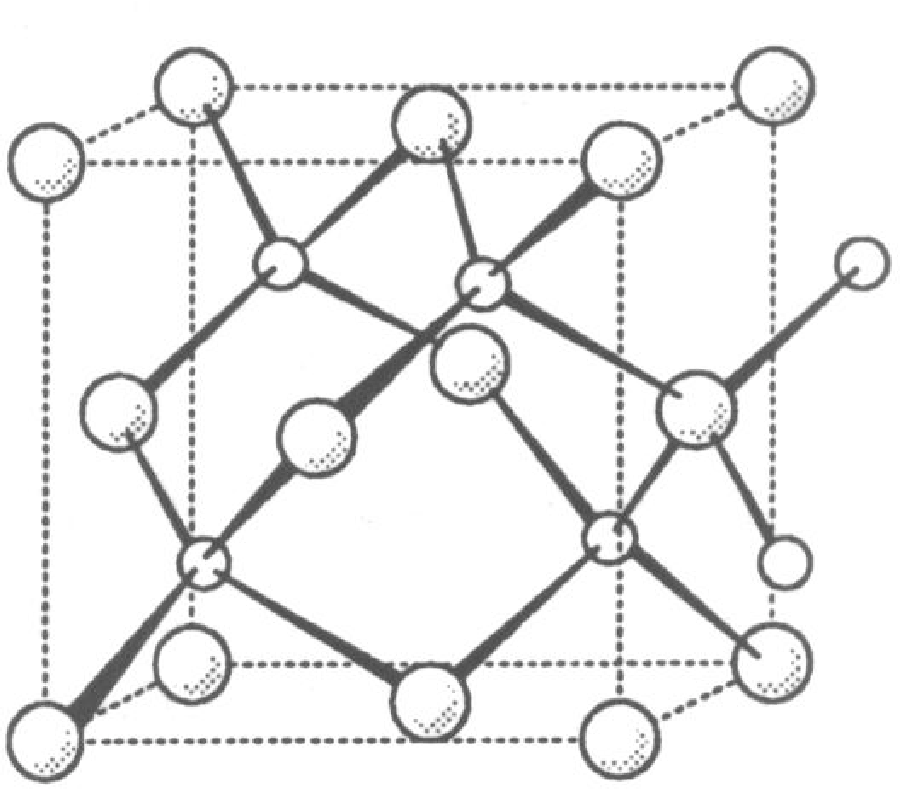
|
|---|---|---|
Wszystkie te materiały krystalizują w strukturze typu wurcytu (rys. 5.1a), typu sfalerytu11 1 Struktura sfalerytu, czyli kubiczna nazywana jest również strukturą regularną. Nazwy “kubiczna” i “regularna” są równoważne i stosowane w krystalografii równie często. W tej pracy będziemy używać pierwszej z nich. (rys. 5.1b), obu tych strukturach lub ich mieszaninie, tworząc kryształy z jednowymiarowym nieuporządkowaniem. Parametry sieci , i zestawiono w tabeli 5.1.
| Struktura | kubiczna | hexagonalna | 1 warstwa | ||
|---|---|---|---|---|---|
| 4.3596 | 3.07521 | 5.048 | 3.07521 | 2.5109 | |
| 3.56683 | 2.5221 | 4.119 | 2.5221 | 2.05931 | |
| 4.5027 | 3.189 | 5.186 | 3.189 | 2.595 | |
5.1.1.1 Węglik krzemu SiC
Węglik krzemu krystalizuje w strukturze blendy cynkowej, rys. 5.1a, (nazywanej też: “typ ”, “struktura wurcytu”) oraz w wielu jej politypowych modyfikacjach. Najczęściej spotykana jest odmiana politypowa (6-cio warstwowa komórka elementarna o symetrii hexagonalnej), zwana , która powstaje podczas gorącej syntezy z pierwiastków. W warunkach wysokiego ciśnienia i temperatury krystalizuje w postaci , struktury typu sfalerytu (rys. 5.1b,c). Jest to struktura o symetrii regularnej (znana też jako: “kubiczna”, “struktura sfalerytu”). Znany jest również cały szereg politypowych struktur , np. , , , , , , , [49]. Węglik krzemu wyjątkowo krystalizuje także w strukturze , czyli wurcytu. Nanokryształy węglika krzemu zostały znalezione w meteorycie Murchison’a [5].
Mikrometrowe polikryształy (rys. 5.6) otrzymuje się najczęściej na drodze bezpośredniej syntezy z pierwiastków (tzw. metoda ). Reakcja chemiczna mieszaniny rozdrobnionego węgla i krzemu:
przebiega w ciśnieniu atmosferycznym, jest silnie egzotermiczna i niekontrolowana (tj. przebiega gwałtownie aż do przereagowania wszystkich substratów) - stąd brak możliwości kontroli rozmiaru otrzymywanych krystalitów.
Węglik krzemu można otrzymać też w reakcji metanu z silanem (). Płomień metanu w atmosferze silanu wytwarza duże ilości ciepła, gazowy wodór oraz nanokrystaliczny o dużej czystości (poniżej wolnego węgla i krzemu). Tą metodą zostały zsyntetyzowane próbki o symbolach k1, k2, k3 (rys. 5.7), k6 i k8 [20].
 |
 |
Kolejna technika polega na pirolizie związków krzemoorganicznych w kontrolowanych temperaturach. W temperaturze około °C następuje rozkład organicznego polimeru i utworzenie sieci nieorganicznych molekuł, tzw. amorficznej ceramiki kowalentnej (, amorphous covalent ceramics), fazy podobnej do szkła. Następnie przez wygrzewanie w temperaturach od do °C prowadzi się do kontrolowanego rozrostu ziaren . Dobranie odpowiednich temperatur i czasów wygrzewania pozwala na otrzymanie nanokryształów o pożądanych rozmiarach (rys. 5.2). Materiał jest bardziej zanieczyszczony niż ten otrzymywany z płomienia, ale dzięki precyzyjnie kontrolowanym warunkom wzrostu ziarna mają mniejszy rozrzut wielkości. Tą metodą otrzymano próbki o symbolach 157k (rys. 5.2), ew3k i h1k [25].
5.1.1.2 Diament
Mikrometrowe i większe kryształy diamentu krystalizują w strukturze kubicznej i są klasycznym jej przykładem (“struktura diamentu”). Komórka elementarna diamentu jest identyczna z komórką , w której wymieniono wszystkie atomy krzemu na węgle, lub z komórką z rys. 5.1c (po zamianie i ). Jest to struktura o bardzo wysokiej symetrii, dlatego daje nieliczne linie dyfrakcyjne, z których pierwsza przypada na dość odległą wartość wektora rozpraszania Å. Kubiczny diament nanokrystaliczny posiada często błędy ułożenia, co upodabnia jego strukturę do hexagonalnej.
 |
 |
Diament otrzymuje się z innych odmian węgla (np. z grafitu) w warunkach wysokiego ciśnienia i temperatury, często z dodatkami metali katalizującymi wzrost jego kryształów. Wysokociśnieniowe procesy syntezy diamentu mogą być w ogólności równowagowe (kwazistatyczne) lub nierównowagowe (dynamiczne). Do tych pierwszych zalicza się syntezę w prasach wysokociśnieniowych (np. toroidalnych) trwającą zazwyczaj godziny. Do tych drugich należy synteza nanokryształów diamentu podczas eksplozji odpowiednio przygotowanego materiału wybuchowego, rys. 5.3. Ten drugi proces trwa mikrosekundy i w odróżnieniu od pierwszego nie zapewnia jednolitych warunków ciśnienia i temperatury w całej objętości próbki. Istnieją dwie odmiany syntezy wybuchowej diamentu:
-
•
Tworzy się on z węgla zawartego w samym materiale wybuchowym. W tym przypadku diament syntezuje w całej objętości komory, w której następuje eksplozja. Ma postać luźnych (niezaglomerowanych) kryształów. W ten sposób zostały zsyntetyzowane próbki dia16, kl5 i kl30.
-
•
Diament tworzy się w wyniku przemiany fazowej grafitu w warunkach eksplozji. Tą techniką można otrzymać polikryształy zarówno nanometrowe jak i większe (nawet do mikrometrów). Polikryształy diamentu otrzymywane tą drogą są zaglomerowane i spieczone. Zawierają dużo błędów ułożenia. Próbka nazywana “dalan” jest komercyjnie dostępnym materiałem otrzymywanym tą techniką.
5.1.1.3 Azotek galu GaN
Stabilną fazą krystalograficzną objętościowych kryształów jest w warunkach normalnych struktura wurcytu, nazywana też politypem (rys. 5.1a). Wysokociśnieniową modyfikacją powyżej jest struktura soli kamiennej [52] (rys. 5.1b). Przy pomocy metod amonotermalnych (reakcja galu z amoniakiem w obecności soli amonu i odpowiedniej temperaturze) udało się otrzymać o strukturze czysto kubicznej [46][19], jednak standardowo mikrometrowe kryształy rosną w strukturze ze sporadycznie występującymi błędami ułożenia. Wraz ze zmniejszaniem rozmiaru ziarna, poniżej obserwuje się zwiększanie liczby błędów ułożenia w tej strukturze.
 |
 |
5.1.2 Politypia
Zjawisko politypii zostało odkryte 90 lat temu. W roku 1912 Baumhauer [2] opisał zdolność materiału do krystalizowania w postaci struktur różniących się tylko w jednym kierunku krystalograficznym.
5.1.2.1 Warstwy politypowe
Pojedyncza warstwa leżących w jednej płaszczyźnie atomów lub cząsteczek, z których zbudowany jest kryształ, nazywana jest monowarstwą, monowarstwą molekularną lub po prostu warstwą. Np. węglik krzemu zbudowany jest z warstw zawierających pary , leżące w płaszczyźnie , jak na rys. 5.5b. Monowarstwa diamentu składa się z par atomów węgla; azotku galu - z par , itd.
a)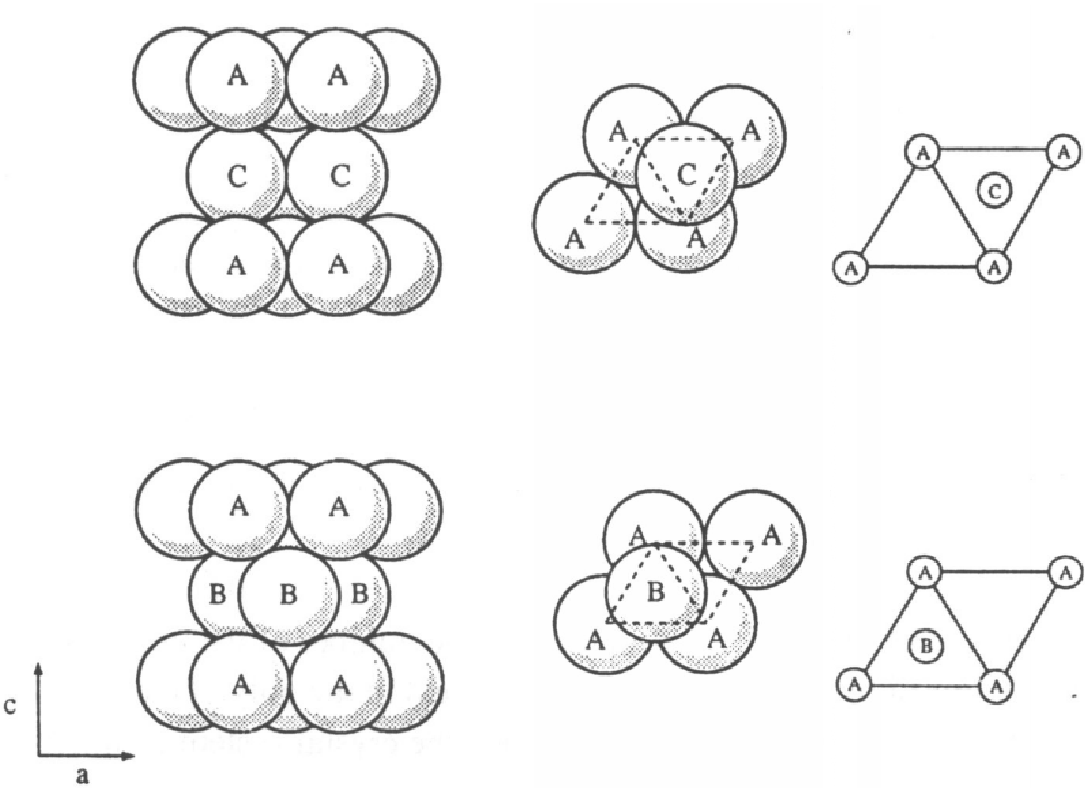 b)
b)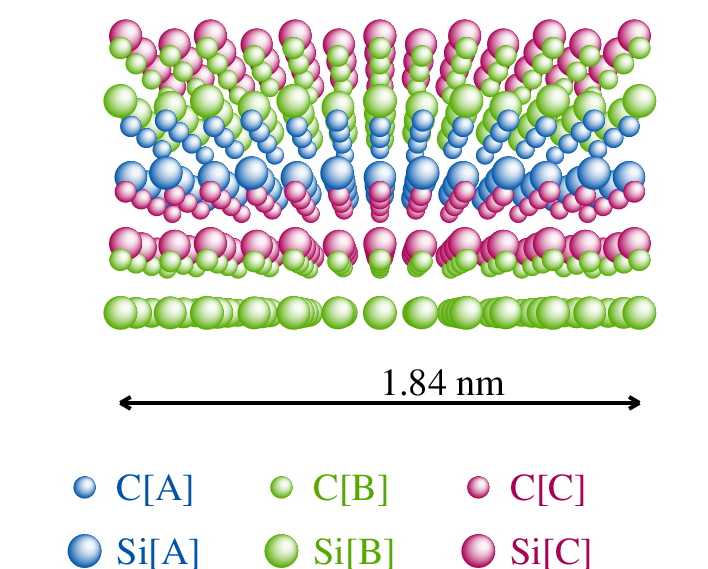
W kryształach o symetrii hexagonalnej (np. , , ) układane na sobie kolejno warstwy mogą przyjmować jedną z trzech pozycji względem wybranego układu współrzędnych (rys. 5.5a). Pozycje te nazywane są tradycyjnie A, B i C i określają typ monowarstwy. W hexagonalnym układzie współrzędnych (kąt między osiami i wynosi 120 stopni), wektory przesunięcia płaszczyzn wynoszą:
-
•
A:
-
•
B:
-
•
C: ,
gdzie jest parametrem sieci.
W strukturze najgęstszego upakowania kolejne warstwy atomów lub cząsteczek, układane kolejno na sobie, tworzą sekwencję politypową (stacking sequence), rys. 5.5a. Najprostszą formą opisu sekwencji warstw jest podanie ciągu liter A,B lub C odpowiadających pozycjom kolejnych warstw. Notacja ta, nazywana notacją ABC, jest najogólniejsza i jednoznacznie opisuje każdą sekwencje warstw, włączając układy całkowicie nieuporządkowane.
Notacja Jagodzińskiego (notacja ) [15][16][17] dzieli wszystkie warstwy na dwa rodzaje: hexagonalne () i kubiczne (). Warstwa jest hexagonalna jeśli obie sąsiadujące z nią warstwy są w jednakowych pozycjach: AA, BB lub CC. Warstwa jest kubiczna jeśli obie sąsiadujące z nią warstwy są w różnych pozycjach: AC, BA lub CA. Notacja opisuje sekwencję politypową jako ciąg warstw i , np.:
-
•
ABCABCABACBACBA w notacji odpowiada:
-
•
xcccccchccccccx w notacji .
-
•
ABABABABCBCBCBC w notacji odpowiada:
-
•
xhhhhhhchhhhhhx w notacji .
W notacji skrajne warstwy () są nieokreślone z braku jednego z sąsiadów. Zaletą notacji Jagodzińskiego jest czytelny zapis obecności (lub braku) błędów ułożenia. Kryształowi czysto kubicznemu odpowiada sekwencja , zaś każdy błąd ułożenia pojawia się w niej jako wtrącenie litery , np. . W krysztale hexagonalnym (sekwencja ) błąd ułożenia to wtrącenie warstwy kubicznej, np. . W kryształach nieuporządkowanych mówi się o rozkładach długości domen hexagonalnych i kubicznych, które są reprezentowane w notacji Jagodzińskiego przez sekwencje warstw i .
Okres politypowy jest to najmniejsza liczba warstw potrzebna do zbudowania politypu (poprzez ich powielanie).
5.1.2.2 Błędy ułożenia
Kryształy z błędami ułożenia to takie, których okres politypowy przekracza fizyczny rozmiar kryształu [21]. Jednowymiarowa (super)struktura politypowa nie powtarza się wtedy w krysztale, a więc nie bierze udziału w konstruktywnej interferencji promieni X. W przypadku kryształów nanometrowych wielkość krystalitu jest porównywalna z okresem politypowym (np. okres politypu wynosi około ). Jednocześnie łamana przez błędy ułożenia symetria kryształu silnie wpływa na zjawisko dyfrakcji. Stwierdzono również, że uporządkowanie dalekiego zasięgu, włączając politypię, zależy od wielkości ziarna (np. nanokryształy diamentu zawierają błędy ułożenia podczas gdy większe są od nich wolne).
Błędy ułożenia są natury stochastycznej, dlatego mogą być opisane przez prawdopodobieństwo ich zaistnienia. Prawdopodobieństwo błędu ułożenia można zdefiniować jako:
-
•
prawdopodobieństwo znalezienia warstwy w strukturach typu sfalerytu:
-
•
prawdopodobieństwo znalezienia warstwy w strukturach typu wurcytu:
Ponieważ , do opisu błędów ułożenia wystarczy zaangażować tylko (tak zwany parametr hexagonalności).
5.1.3 Mikrostruktura polikryształów nanometrowych
W niniejszej pracy pod pojęciem mikrostruktury proszku będziemy rozumieli:
- jego morfologię
-
w sensie mikroskopowym, czyli cechy krystalitów rozumianych jako kawałki materii, z zaniedbaniem ich struktury atomowej, np.
-
•
wielkość lub rozkład wielkości krystalitów (ziaren),
-
•
kształt ziaren,
-
•
wzajemne ułożenie ziaren, np. przypadkowe lub posiadające jakieś prawidłowości, superstrukturę.
- makroskopowe zaburzenia (defekty) struktury atomowej
-
krystalitów w stosunku do ich sieci zrelaksowanej, które wpływają na ich kształt czy orientację ziaren, np.
-
•
naprężenia sieci krystalicznej,
-
•
defekty sieci krystalicznej,
-
•
błędy ułożenia.
Morfologia proszku i zaburzenia struktury atomowej są ze sobą powiązane, np. w eksperymentach wysokociśnieniowych układ ziaren w proszku może implikować określone zmiany naprężeń krystalitów. I odwrotnie: wywołany naprężeniami ruch dyslokacji zmienia kształt (a czasem i rozmiar) ziaren. Nie będą dalej dyskutowane przyczyny defektów sieci krystalicznej i mechanizmy ich powstawania lecz niektóre ich skutki widoczne w postaci deformacji linii dyfrakcyjnych.
W opisie mikrostruktury używa się m.in. pojęć:
- krystalit, ziarno:
-
pierwotny, najmniejszy element proszku. Tutaj przyjmiemy, że jest nim nanokryształ jednolity pod względem struktury atomowej.
- aglomerat:
-
układ (zlepek) wielu ziaren. Ziarna aglomeratu przylegają do siebie na tyle ściśle, że ich powierzchnia swobodna jest mniejsza niż izolowanych ziaren. Do aglomeracji ziaren dochodzi np. w procesach zagęszczania nanoproszków. Aglomeraty ziaren nanometrowych często mają postać fraktali.
5.1.3.1 Ułożenie krystalitów w proszku
Przestrzenne ułożenie krystalitów w proszku zdeterminowane jest przez siły ich wzajemnego oddziaływania. Może to być zarówno przyciąganie elektrostatyczne jak siły Van der Waals’a czy - w przypadku gęstych ceramik (spieków) - wiązania kowalencyjne, jonowe lub inne (w zależności od rodzaju materiału). Siły wiążące ziarna proszku (siły adhezji), bez względu na ich rodzaj, działają pomiędzy atomami przypowierzchniowymi i są do wielkości tych powierzchni proporcjonalne. Z drugiej strony siły prowadzące do rozseparowania ziaren, jak udary mechaniczne, czy chociażby grawitacja, rosną z momentem bezwładności i masą ziaren, a więc z ich objętością. W tej sytuacji stabilność mechaniczna proszku podyktowana jest w dużej mierze stosunkiem sił wiążących (adhezji) do sił separujących, a więc i stosunkiem powierzchni krystalitów do ich objętości [14]:
| (5.1) |
gdzie jest objętością materiału zgromadzonego nie dalej niż o od powierzchni ziarna w kształcie kuli o średnicy i objętości . można traktować jako obszar granic pomiędzy ziarnami, zaś wytrzymałość mechaniczna proszku jest proporcjonalna do . Jak widać, wytrzymałość maleje jak co stanowi podstawę do podziału proszków na dwie klasy:
-
•
“zwykłych”, czyli mikrometrowych, gdzie duża masa ziaren wobec słabo rozwiniętej powierzchni uniemożliwia tworzenie stabilnych struktur przestrzennych (rys. 5.6a)
- •
Ułożenie przypadkowe - proszki mikrometrowe
Ten rodzaj ułożenia jest charakterystyczny dla proszków złożonych ze stosunkowo dużych ziaren (rzędu i więcej).
Siły adhezji działające na niewielkiej powierzchni styku ziaren są zbyt słabe aby stworzyć z dużych kryształów trwałą superstrukturę (rys. 5.6). Proszek zachowuje się w sposób podobny do sypkiego piasku lub cieczy: gęsto wypełnia objętość którą ma do dyspozycji, rys. 5.6a.
a)
|
b)
|
Wypełnienie objętości przez ziarna ułożone przypadkowo jest duże (do ).
Ułożenia łańcuchowe i fraktalne - proszki nanometrowe
Bardzo małe kryształy (kilka i kilkanaście ) wykazują tendencję do tworzenia superstruktur przestrzennych w trakcie ich syntezy. Duży stosunek powierzchni (a więc sił adhezji) do masy krystalitu zapewnia znaczną stabilność takich superstruktur. Mogą one mieć postać rozgałęzionych włókien, często tworzą struktury fraktalne o znacznej rozpiętości masowych wymiarów fraktalnych (od wzwyż), rys. 5.7.
a)
b)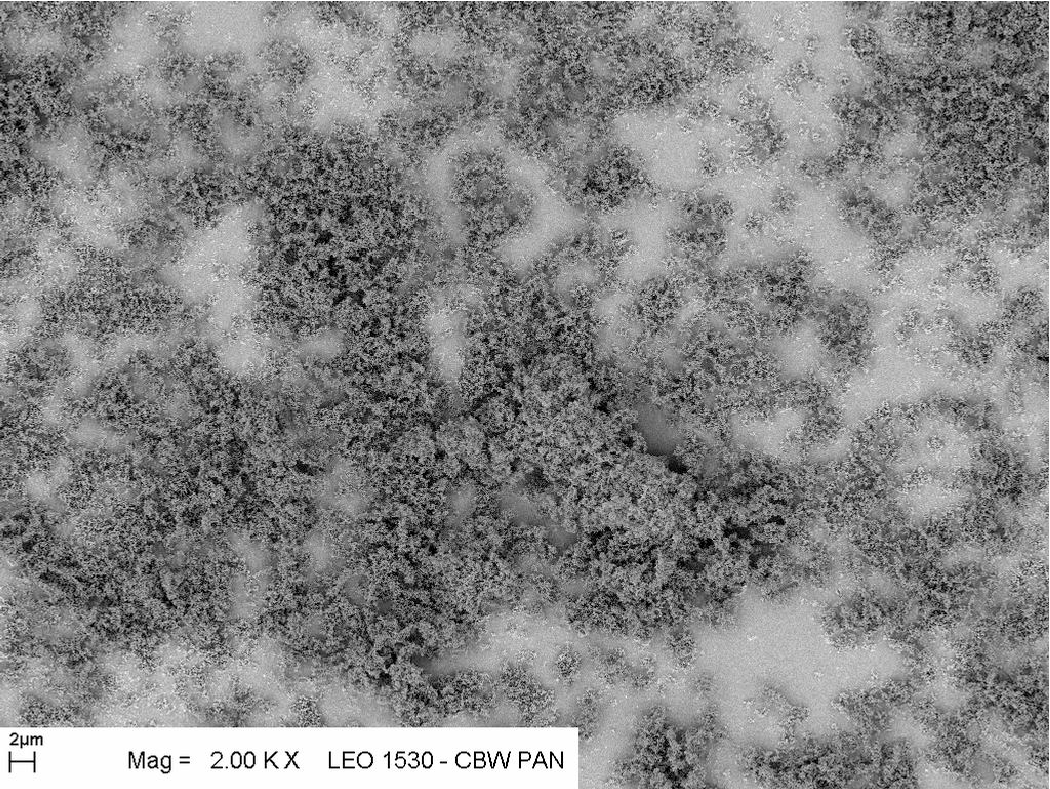 c)
c)
d) e)
e)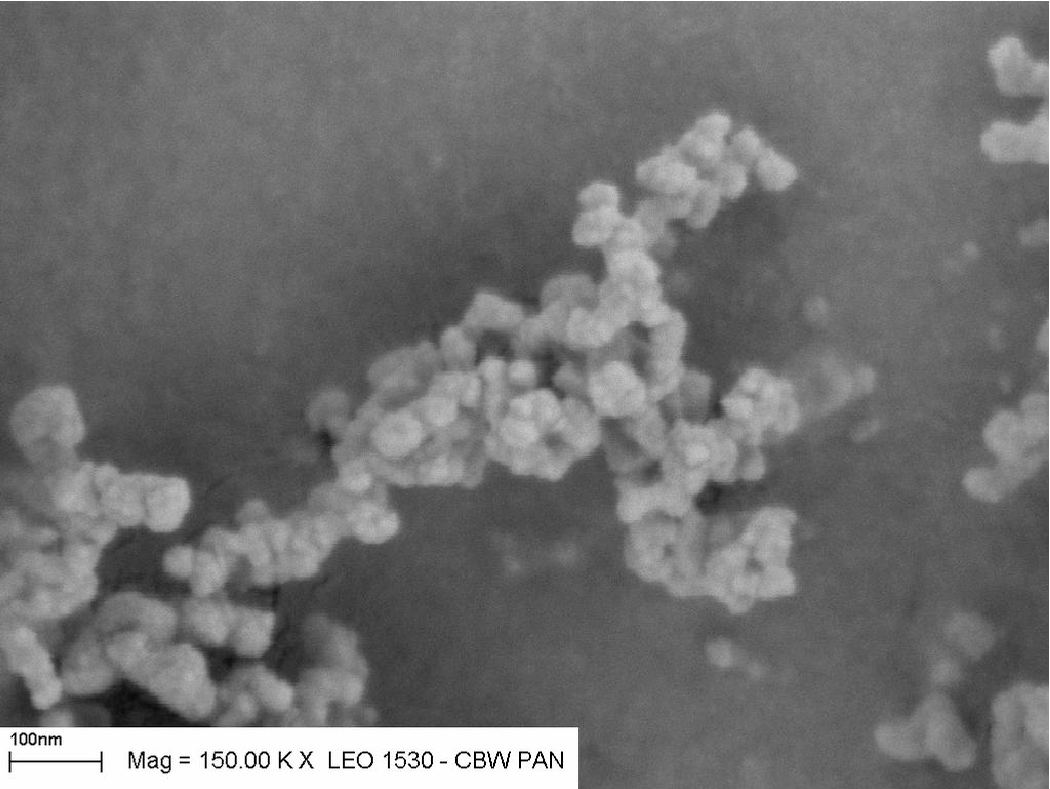
Istnieje uporządkowanie dalekiego zasięgu (tj. poza drugich sąsiadów) ziaren. Odległości międzyatomowe będące wielokrotnością średnicy ziarna są bardziej prawdopodobne niż inne. Sytuacja ta nie ma analogii dla struktury atomowej.
Ułożenie gęste - ceramika
- Ceramika
-
jest wytrzymałym materiałem nieorganicznym i niemetalicznym. Do zastosowań praktycznych używa się materiały takie jak węglik krzemu (), czyste tlenki (), azotki (), szkła niekrzemianowe i wiele innych. Ceramiki są twardsze i sztywniejsze od stali, bardziej odporne na wysoką temperaturę i korozję niż metale i polimery, lżejsze od wielu metali i ich stopów.
Ceramika jest granicznym stanem zagęszczenia proszku złożonego z kryształów. Idealna ceramika ma gęstość zbliżoną do gęstości materiału z którego jest zbudowana. Krystality stykają się ze swoimi sąsiadami całą swoją powierzchnią, wykorzystując do maksimum siły adhezji. Tworzą się wiązania chemiczne. Ziarna zachowują jednak odrębność własnych struktur krystalicznych, co utrudnia m.in. propagację dyslokacji pomiędzy nimi.
5.1.4 Metody doświadczalne w badaniu mikrostruktury
W pracy niniejszej korzystano z czterech metod doświadczalnych pomocnych w badaniach mikrostruktury: mikroskopii elektronowej (Scanning Electron Microscopy ), pomiarów powierzchni właściwej (Specific Surface ), proszkowej dyfrakcji rentgenowskiej (X-Ray Diffraction ) i niskokątowego rozpraszania promieni rentgenowskich i neutronów (Small Angle Scattering ). Dwie pierwsze metody dostarczają wyników samowyjaśniających (fotografie, wartość powierzchni właściwej) i nie będą dokładniej omawiane. O obu zaś metodach dyfrakcyjnych trzeba powiedzieć, że są komplementarne i razem dają komplet informacji mikrostrukturalnej:
-
•
dyfrakcja proszkowa w zakresie wysokich kątów (tzw. części bragowskiej widma) pozwala obserwować strukturę atomową kryształów i jej zaburzenia. Warunki brzegowe dyfrakcji (dystrybucja kształtu kryształu) przejawiające się w postaci profilu linii dyfrakcyjnej pozwalają też na dokładny pomiar rozkładu wielkości krystalitów w proszku.
-
•
rozpraszanie niskokątowe pozwala określać przestrzenny rozkład krystalitów, ich rozmiary oraz aglomerację. Rozpraszanie to odbywa się na granicach między ziarnami a otoczeniem.
5.1.4.1 Dyfrakcja proszkowa
Rozpraszanie promieni rentgenowskich (lub neutronów22 2 Promienie X i neutrony mogą być stosowane zamiennie we wszystkich opisanych tu przypadkach. ) na proszkach złożonych z nanokryształów jest bodaj najprostszą i najbardziej efektywną metodą badań struktury atomowej i rozkładu wielkości krystalitów w tych materiałach. W badaniach tych korzysta się z dyfraktometrów proszkowych, mogących pracować w dwóch geometriach:
-
•
z rozdzielczością kąta (angle dispersive, ruchomy detektor przy monochromatycznej wiązce padającej)
-
•
z rozdzielczością energii (energy dispersive, nieruchomy detektor przy polichromatycznej wiązce padającej)
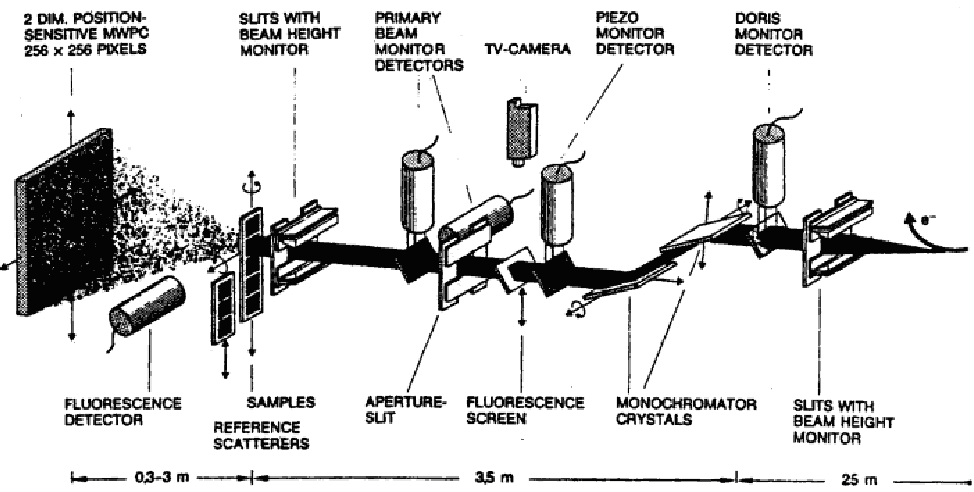
Pierwszy układ jest bardziej rozpowszechniony, gdyż laboratoryjne źródła promieni X (lampy rentgenowskie) emitują promieniowanie o określonych długościach fali, charakterystycznych dla materiału anody. Układ z rozdzielczością kąta pozwala na pomiar danych o wysokiej rozdzielczości i dużym stosunku sygnału do szumów. Stosunek ten można dodatkowo poprawić stosując synchrotronowe źródło promieni X.
a)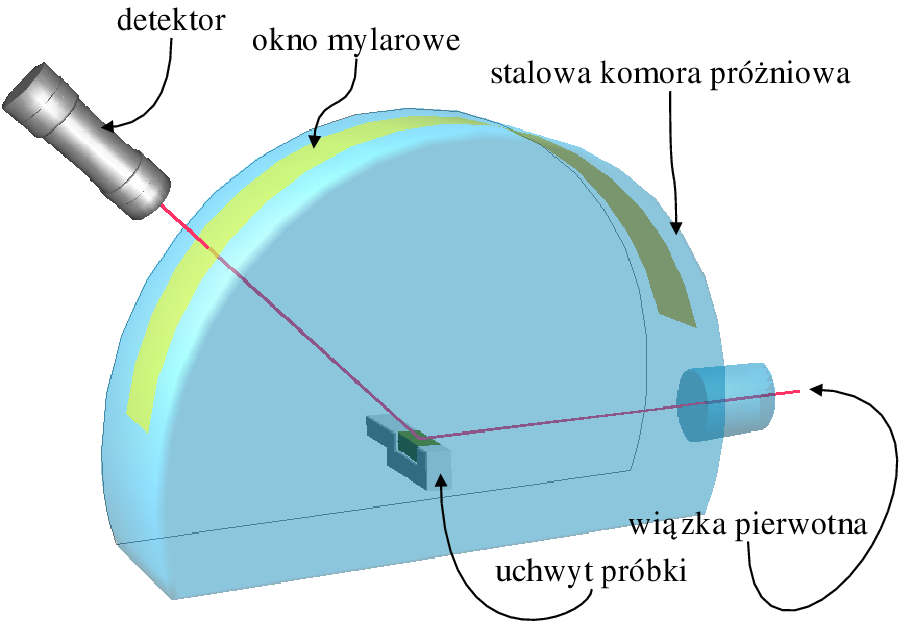
|
b)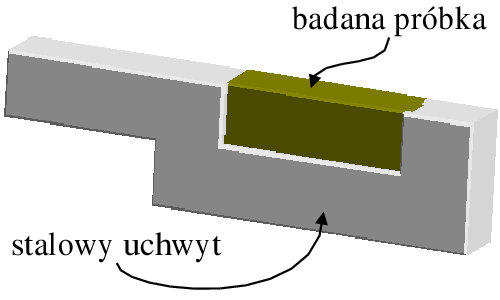
|
Krzywe dyfrakcyjne nanoproszków - w porównaniu do polikryształów mikrometrowych - mają bardzo szerokie maksima, a więc relatywnie mniejsze natężenia w tych maksimach i w rezultacie - mniejszy stosunek sygnału do szumu. W związku z tym nawet niewielkie ograniczenie źródeł szumów i zakłóceń w przypadku nanoproszków zdecydowanie poprawia jakość danych. Celem zlikwidowania źródeł szumu można umieścić wiązkę padającą, próbkę oraz wiązkę rozproszoną w próżni. Zapobiega to rozpraszaniu i absorpcji w powietrzu (rys. 5.8a). Próżniowy układ dyfrakcyjny, konstrukcji CBW PAN, pracuje na linii B2 laboratorium HASYLAB synchrotronu DESY w Hamburgu. Dodatkowo posiada on specjalnie zaprojektowany uchwyt do próbek (rys. 5.8b), który eliminuje używaną zazwyczaj szklaną kapilarę i w połączeniu z układem próżniowym gwarantuje, że fotony mają kontakt wyłącznie z próbką i detektorem. Ten unikalny układ pomiarowy pozwala na obserwację słabych refleksów (normalnie ukrytych w tle) i ilościowy pomiar rozpraszania dyfuzyjnego próbki. Układ pracuje w geometrii Debye’a-Scherrer’a.
Układ z rozdzielczością energii nie posiada wymienionych zalet, ale jest użyteczny w wysokociśnieniowych (i nie tylko) doświadczeniach in situ, gdzie aparatura wytwarzająca pożądane warunki eksperymentalne (np. prasa wysokociśnieniowa) szczelnie otacza próbkę i pozostawia niewielkie światło dla rozproszonego promieniowania. W geometrii z rozdzielczością energii do wykonania pomiaru wystarczy wąska szczelina w aparaturze. Ta geometria stosowana jest najczęściej na synchrotronach i źródłach neutronowych (z powodu białego promieniowania i możliwości przeniknięcia silnej wiązki przez warstwy materiału otaczającego próbkę).
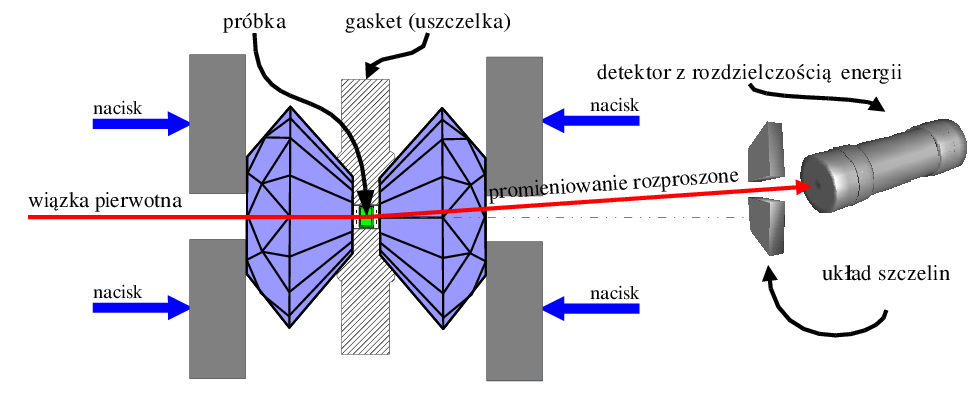
Przykładem zastosowania układu z rozdzielczością energii jest wysokociśnieniowy pomiar dyfrakcyjny in situ w kowadłach diamentowych (rys. 5.9). Próbka umieszczona jest w niewielkim otworze wykonanym w blaszce (gaskecie). Dwa monokryształy diamentu, umocowane w mechanicznym zacisku (dokręcanym śrubą) wywierają nacisk na gasket i próbkę. Przez otwory w zacisku przechodzi wiązka pierwotna. Przechodzi ona przez pierwszy monokryształ diamentu (ustawiony w położeniu nieodbijającym), następnie przez próbkę i ulega tam rozproszeniu. Promieniowanie rozproszone wychodzi z układu przez drugi monokryształ i jest rejestrowane w nieruchomym detektorze, który mierzy ilość kwantów i ich energię. Otrzymany w ten sposób dyfraktogram jest funkcją natężenia od energii (a nie kąta). Ciśnienie możliwe do uzyskania w kowadłach diamentowych sięga , jednak typowe warunki pracy nie przekroczyły w przypadku opisanych pomiarów . Pomiaru przyłożonego ciśnienia dokonuje się poprzez obliczenie parametru sieci wzorcowej domieszki (wskaźnika ciśnienia), której dodaje się do badanego materiału, np. , .
5.1.4.2 Rozpraszanie niskokątowe ()
Rozpraszanie niskokątowe (Small Angle Scattering, ) promieni rentgenowskich (Small Angle X-Ray Scattering, ) lub neutronów (Small Angle Neutron Scattering, ) jest metodą pozwalającą badać mikrostrukturę obiektów o rozmiarach do kilkudziesięciu . W odróżnieniu od dyfrakcji wysokokątowej, obiektami rozpraszającymi są tu nie atomy a całe ziarna obecne w proszku. nie jest więc czuły na strukturę atomową - pokazuje budowę nanomateriału, średnie relacje geometryczne pomiędzy ziarnami (na co nie jest czuła dyfrakcja wysokokątowa). Eksperymenty są w istocie identyczne z doświadczeniami dyfrakcji wysokokątowej (rys. 5.10), z tą tylko różnicą, że w mierzymy tylko jedną linię dyfrakcyjną (o indeksie ) i w konstruktywnej interferencji biorą udział wszystkie atomy próbki - stąd natężenia ogromne w porównaniu z maksimami bragowskimi. Specyfiką pomiaru linii jest obecność w mierzonej wiązce obok promieniowania rozproszonego na próbce, także nierozproszonej wiązki pierwotnej (promieniowania przechodzącego bez oddziaływania z próbką). Obszar kątowy, w którym oba strumienie fotonów się mieszają jest bezużyteczny i zatrzymuje się go na chwytce. Ogranicza to od dołu najmniejszy osiągalny pomiarowo kąt ugięcia33 3 Problemów technicznych przy pomiarach jest oczywiście więcej. Główne to rozpraszanie na powietrzu i krawędziach szczelin definiujących wiązkę.. Doświadczenia są szczególnie użyteczne przy badaniach zagęszczania proszków nanokrystalicznych. Z racji opisanych ograniczeń (najlepsze stacje pozwalają mierzyć natężenia ugięte pod kątami odpowiadającymi wektorowi rozpraszania nie mniejszemu od Å, rys. 5.10), w praktyce można określić średnie otocznie krystalitów proszku nie dalej niż na odległość sąsiadów.
5.2 Badania nanokryształów metodą ab initio
Nanokryształy o strukturze najgęstszego upakowania zawierające błędy ułożenia stanowią szczególnie trudny obiekt przy próbach charakteryzacji ich struktury atomowej i mikrostruktury metodami dyfrakcyjnymi. O ile w kryształach mikrometrowych istnienie błędów ułożenia nie jest regułą (np. nie ma ich zazwyczaj w i ), o tyle nanokryształy prawie zawsze zawierają znaczną ich liczbę. Problem z błędami ułożenia polega na tym, że tworzą one dodatkowe maksima natężenia położone bardzo blisko refleksów strukturalnych materiału. Dla nanokryształów, których linie dyfrakcyjne są niezwykle szerokie ze względu na rozmiar ziarna, oznacza to poszerzenie i deformację refleksów strukturalnych, co w oczywisty sposób utrudnia, a często wręcz uniemożliwia wyznaczenie ich szerokości i położeń. Trudno jest bowiem rozseparować wkłady wielkości krystalitu i błędów ułożenia w szerokość linii, nie mówiąc o ilościowej analizie jej profilu (rozkład wielkości ziaren). Próby “włączenia” błędów ułożenia do struktury i obliczania zbioru refleksów zawierających również te wynikające z jednowymiarowego nieuporządkowania były podejmowane dla kryształów mikrometrowych [50]. Jednak w przypadku nanokryształów, gdzie mamy najwyżej kilka błędów ułożenia rozłożonych losowo w krystalicie (ma on kilkanaście warstw atomów), trudno stworzyć jeden model struktury kryształu, skoro mamy do czynienia z mieszaniną ogromnej ilości kryształów, z których każdy ma inną strukturę krystaliczną, dającą (z osobna) inny obraz dyfrakcyjny.
Skoro trudno jest oddzielić w dyfraktogramie efekty pochodzące od rozmiaru i błędów ułożenia, dobrym wyjściem jest podejście odwrotne: raczej składanie niż rozdzielanie. Można bowiem obliczyć dyfraktogram ab initio tak, aby zawierał oba wspomniane komponenty i spróbować dopasować ich relacje tak, żeby osiągnąć zgodność z danymi doświadczalnymi [42].
5.2.1 Analiza danych doświadczalnych metodą obliczeń ab initio
Przy pomocy metod obliczeniowych opisanych w rozdziale 4.2, w szczególności korzystając z uśredniania jednowymiarowego nieuporządkowania z użyciem wielowarstwowej funkcji rozkładu radialnego (§4.2.4), można obliczyć dyfraktogram nanoproszku złożonego z krystalitów o różnych wielkościach () i jednocześnie zawierających błędy ułożenia zdefiniowane statystycznie poprzez prawdopodobieństwo ich wystąpienia.
Obliczenia dokonywane są przez program rpnano44 4 Pakiet rpnano dostępny jest pod adresem http://dev.pielaszek.net/public/nar/. Problemy i pytania prosimy zgłaszać pod adresem support@science24.com., będący implementacją algorytmów opisanych w rozdziale 4.2. Stosowany rozkład wielkości ziaren jest log-normalny (por. paragraf 4.2.3) i posiada dwa parametry: położenie maksimum i dyspersję . Trzecią zmienną jest parametr hexagonalności, , będący prawdopodobieństwem znalezienia warstwy typu w krysztale (por. 4.2.4). Po ustaleniu wartości powyższych trzech parametrów55 5 Niekiedy konieczne jest dodatkowo podanie wartości czynnika temperaturowego . obliczany jest metodą ab initio dyfraktogram proszkowy badanego materiału. Trwa to od jednej do dwóch sekund. Opisany pojedynczy cykl obliczeniowy zamknięty jest w pętli kontrolowanej przez algorytm genetyczny.
5.2.1.1 Algorytm genetyczny
Zadaniem algorytmu genetycznego [47] używanego do sterowania przebiegiem obliczeń ab initio jest takie manipulowanie wartościami parametrów , i , żeby osiągnąć jak najlepszą zgodność obliczanych na ich podstawie dyfraktogramów z charakteryzowanymi krzywymi doświadczalnymi. Algorytm genetyczny buduje populację osobników (trójek parametrów , i ), oblicza ab initio odpowiadających im dyfraktogramów i wylicza dopasowania obliczonych modeli do danych doświadczalnych. Następnie z całej populacji wybiera się osobniki najlepiej dopasowane (trójki , i najwierniej odwzorowujące dane doświadczalne) i krzyżuje się je ze sobą. Krzyżowanie polega na częściowej wymianie “informacji genetycznej” (np. dwa osobniki wymieniają się między sobą wartościami parametru ). Następuje też mutacja, polegająca na losowej zmianie wartości losowo wybranych parametrów. Jednak prawdopodobieństwo mutacji jest małe () zaś krzyżowania duże (). Tak utworzone nowe pokolenie przechodzi tą samą procedurę co ich rodzice (wylicza się funkcje dopasowania, dokonuje selekcji, krzyżuje, mutuje, itd.).
W każdym nowym pokoleniu część osobników na krzyżowaniu i mutacjach zyskuje, a część traci. Osobniki lepiej dostosowane przetrwają kolejną selekcję i przeniosą dobrze pasujące wartości , i do kolejnego cyklu, zaś chybione trójki , i znikną bezpotomnie. Stagnacji procesu przeciwdziałają mutacje, które wprowadzają do układu “nowe pomysły” na , i . Dzięki temu algorytm genetyczny jest dość stabilny i nie zatrzymuje się zazwyczaj na lokalnych maksimach dopasowania, nawet przy trudnych problemach optymalizacyjnych, a do takich należy ustalanie wartości trzech silnie skorelowanych parametrów , i .
Przykłady charakteryzacji proszków nanokrystalicznych przy pomocy opisanej metody pokazano na rys. 5.11 i 5.12. Obie krzywe dyfrakcyjne zmierzone zostały na wysokorozdzielczym dyfraktometrze proszkowym linii B2 synchrotronu DESY z użyciem komory próżniowej (por. 5.1.4.1). Krzywa z rys. 5.11a to dyfraktogram nanokrystalicznego otrzymanego metodą pirolizy związków krzemoorganicznych i izotermicznego wygrzewania materiału amorficznego, krzywa z rys. 5.12 to nanometrowy syntezowany w płomieniu, por. 5.1.1.1.
5.2.2 Wyniki
Pierwsza z analizowanych próbek była syntezowana w dwustopniowym procesie. W pierwszym kroku dokonuje się termicznego rozkładu związków krzemoorganicznych w temperaturze °C uzyskując materiał amorficzny. Następnie przez kontrolowane wygrzewanie (°C w czasie ) doprowadza się do nukleacji i wzrostu nanokryształów . Zgodnie z oczekiwaniami rozkład wielkości ziaren takiego materiału jest stosunkowo wąski (patrz rys. 5.11c). Materiał zawiera błędów ułożenia warstw. Funkcja korelacji warstw (rys. 5.11b) pokazuje, że porządek struktury materiału jest zachowany na odcinku (średnio) jedynie 3 warstw. W odległości pięciu warstw od dowolnej warstwy odniesienia krystalitu, znika wszelka korelacja z typem warstw poprzedzających. Oznacza to, że możemy z jednakowym prawdopodobieństwem spotkać tam warstwę w pozycji lub , niezależnie od tego, w jakiej pozycji była warstwa odniesienia. Jest to przypadek bardzo silnego nieuporządkowania struktury najgęstszego upakowania.
Druga z analizowanych próbek była syntezowana przez spalanie silanu w metanie. Rozkład wielkości ziaren otrzymany dla tej próbki jest dwumodowy. Proszek jest mieszaniną dwóch frakcji o rozmiarach ziaren ok. i Å. Frakcja mniejszych krystalitów posiada stosunkowo szeroki rozkład wielkości, zaś krystality większe mają lepiej zdefiniowany rozmiar. Żadna z tradycyjnych metod dyfrakcyjnych nie dostarcza informacji na temat wielomodowych rozkładów wielkości ziaren. W materiale istnieje warstw hexagonalnych (błędów ułożenia). Jest to niewielka różnica w stosunku do próbki pierwszej, jednak porównanie funkcji korelacji warstw obu materiałów (rys. 5.11b i 5.12b) pokazuje, że korelacje pomiędzy pozycjami kolejnych warstw są znacznie silniejsze i zanikając całkowicie dopiero po ok. 10-ciu warstwach. Rozkład wielkości ziaren ma większą szerokość niż poprzedni, co wiąże się z obecnością dwóch frakcji ziaren o różnych rozmiarach.
a)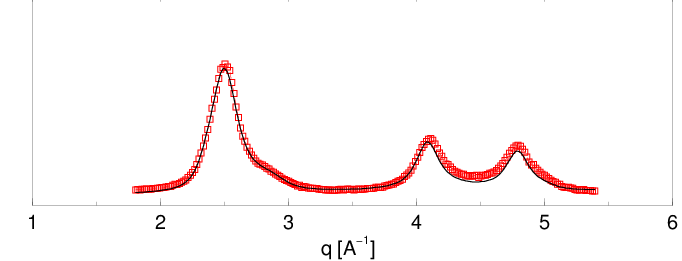
b)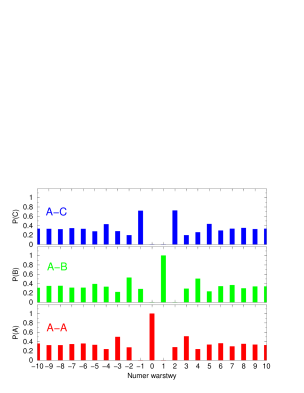 c)
c)
a)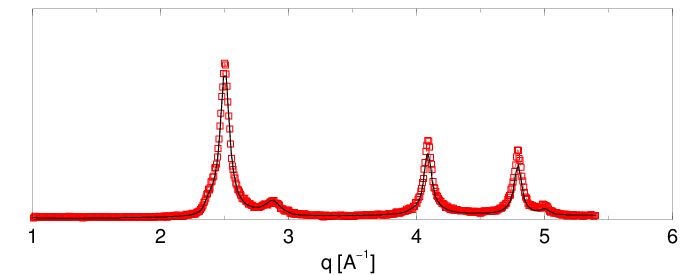
b)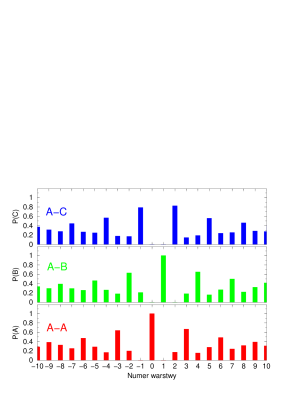 c)
c)
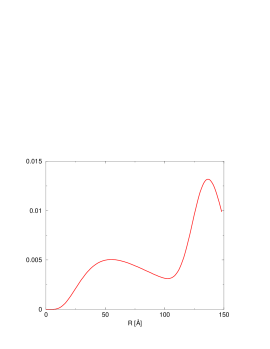
5.2.3 Wnioski
Ilościowe wyznaczenie rozkładu wielkości ziaren proszku nanokrystalicznego jest możliwe nawet w obecności bardzo licznych () błędów ułożenia, pod warunkiem zastosowania silnego narzędzia, jakim są obliczenia dyfrakcji przy wykorzystaniu praw pierwszych (ab initio). Na uwagę zasługuje fakt, iż precyzja oznaczenia rozmiaru krystalitów rośnie wraz ze zmniejszaniem ziarna. Dla materiałów o kryształach mniejszych niż precyzja ta sięga Å, czyli grubości pojedynczej warstwy atomowej. Właśnie z taką precyzją została wyznaczona wielkość krystalitu w próbce otrzymanej z pirolizy związków krzemoorganicznych.
Jak widać z rys. 5.11a i 5.12a, dopasowaniu podlega nie pojedyncza linia dyfrakcyjna a cały dyfraktogram, ponieważ przedstawiony w rozdziale 4.2 formalizm Debye’a w ogóle nie wprowadza pojęć takich jak układ płaszczyzn czy linia dyfrakcyjna (w obliczeniach ab initio operuje się na obiektach takich jak atom, ziarno i proszek). W tej teorii dyfrakcja odbywa się w całej przestrzeni odwrotnej, a nie tylko w węzłach jej sieci jak w przypadku bragowskim.
Takie uogólnienie oprócz oczywistych zalet (bez niego ilościowe dyfrakcyjne badania nanokryształów w ogóle nie byłyby możliwe) ma też wady w postaci znacznej komplikacji obliczeń dyfrakcyjnych i ich dużych kosztów numerycznych. Oprogramowanie powstałe przy okazji tej pracy w praktyce usuwa obie te niedogodności.
Niejako “przy okazji” rozkładu wielkości ziaren otrzymuje się z dopasowania krzywych obliczanych ab initio także statystyczną charakterystykę nieporządku w nanokryształach. Zastosowany model struktury z błędami ułożenia nie narzuca żadnych ograniczeń na miejsca, gdzie błędy pojawiają się w strukturze. Oznacza to brak podziału kryształu na domeny kubiczne i hexagonalne. Jedyną i - jak widać z dopasowań - wystarczającą definicją nieporządku jest prawdopodobieństwo pojawienia się błędu w strukturze. Dla struktur o niewielkiej zawartości błędów ułożenia, powiedzmy do , model domenowy i statystyczny są sobie równoważne, ponieważ sporadycznie pojawiające się warstwy w naturalny sposób dzielą kubiczny kryształ na domeny. Jednak dla dużych koncentracji warstw typu , a z takimi mamy do czynienia w , sukces modelu statystycznego w poprawnym opisie obserwowanej dyfrakcji oznacza brak tendencji warstw do grupowania się. Można powiedzieć, że jest to strukturalna właściwość wynikająca z nanometrowego rozmiaru kryształów, gdyż w mikronowych polikryształach istnieją duże domeny typu wolne od błędów ułożenia oddzielone obszarami silnie nieuporządkowanymi (zawierającymi mieszaninę warstw i ) [50].
Podobnie jak w , błędy ułożenia rozsiane są przypadkowo na całej długości nanokryształów jak również nanokryształów diamentu otrzymywanych z przemiany fazowej grafitu podczas eksplozji (por. §5.1.1.2).
5.3 Wpływ metody syntezy na mikrostrukturę nanokryształów
Rozkład wielkości ziaren (Grain Size Distribution ) stanowi odcisk, ślad po zakończonym procesie syntezy nanokryształów. Analiza procesów termodynamicznych towarzyszących ich wzrostowi wykracza poza ramy niniejszej pracy. Zaprezentowane zostaną jednak fakty doświadczalne świadczące o tym, że względna szerokość jest wartością stałą dla materiałów otrzymanych jedną metodą. Ponadto zostanie oszacowany przedział dyspersji otrzymywanych w czterech rodzajach procesów syntezy (od syntezy izotermicznej po eksplozję materiału wybuchowego).
5.3.1 Wyznaczanie metodą
Do wyznaczenia rozkładu wielkości ziaren nanoproszków i użyto opisanej w §2.3.2 metody . Dla zwiększenia dokładności oznaczeń, do refleksów bragowskich dopasowano funkcję Pearson VII. Do wyznaczenia rozkładu wielkości ziaren wzięto szerokości i dopasowanej funkcji Pearson VII, zgodnie z opisem umieszczonym w §2.3.5.1. W przypadkach linii dyfrakcyjnych zniekształconych przez lokalne maksima pochodzące od błędów ułożenia, wykonano dopasowanie części linii obejmującej kilka punktów pomiarowych wokół maksimum oraz jedno z ramion wolne od zniekształceń.
Ze względu na wymagania jakościowe, do oznaczania dyspersji użyto tylko niektórych zestawów danych i , najczęściej otrzymanych z pomiarów na wysokorozdzielczym dyfraktometrze proszkowym linii B2 synchrotronu DESY w warunkach próżni (paragraf 5.1.4.1). Badane materiały były otrzymywane na trzy sposoby (por. §5.1.1.1 i §5.1.1.2):
-
•
Piroliza związków krzemoorganicznych a następnie izotermiczne wygrzewanie produktów rozkładu (amorficznego ) w temperaturze i ciśnieniu atmosferycznym. Próbki : 157k, ew3k, h1k [25].
-
•
Synteza płomieniowa w ciśnieniu atmosferycznym. Próbki k1, k1c, k1v, k2, k6v, k8 [20].
-
•
Eksplozja w temperaturze ok. i ciśnieniu . Próbki : dalan, kl5, kl30 [proszki diamentowe dostępne komercyjnie].
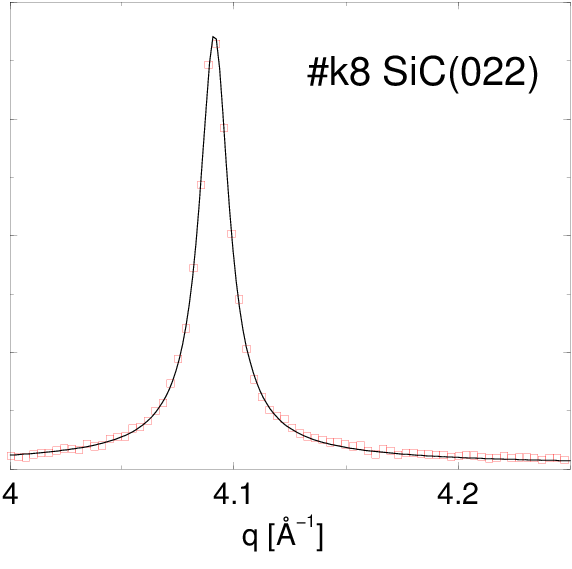
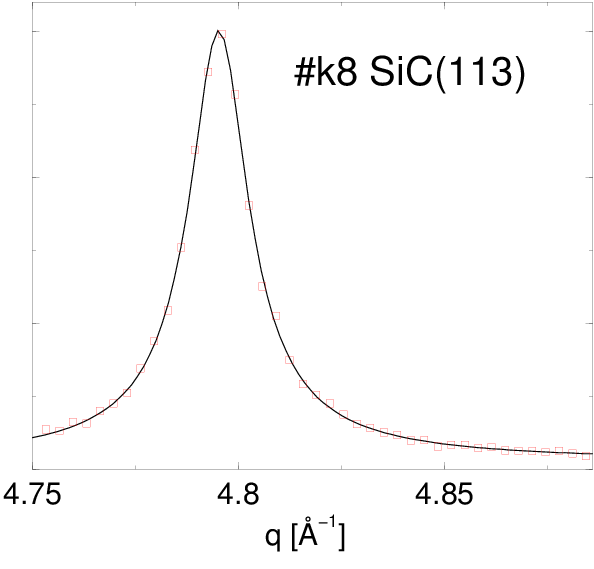
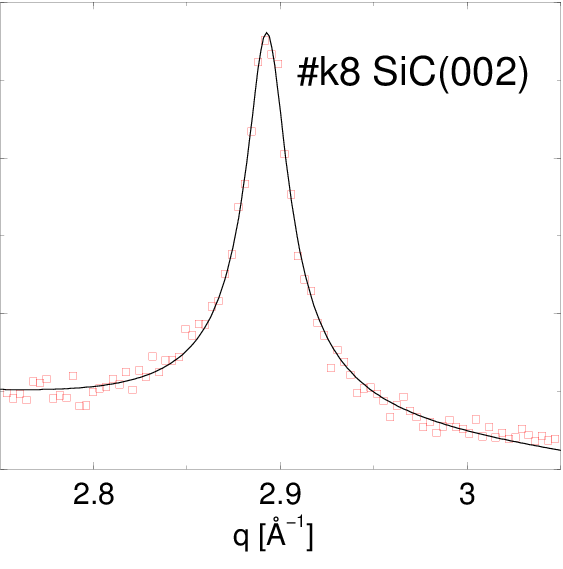
Po dopasowaniu funkcji Pearson VII do najsilniejszych linii dyfrakcyjnych (rys. 5.13), otrzymane parametry i tej krzywej (tabela 5.2) podstawiono do wzorów opisanych w §2.3.5.1 otrzymując średni rozmiar krystalitu w proszku i dyspersję rozkładu wielkości ziaren (tabela 5.2). Można do tego celu użyć m.in. narzędzia internetowego http://science24.com/fw145m/, rys 2.11. Z podzielenia tych wielkości otrzymano wartość “monodyspersyjności”, wielkości charakteryzującej względną szerokość .
 |
 |
 |
5.3.2 Względna szerokość a metoda syntezy
Stwierdzono, że stosunek wyznaczonej doświadczalnie dyspersji do wartości średniej rozkładu wielkości ziaren jest w przybliżeniu stały i charakterystyczny dla miejsca syntezy materiału. Prawidłowość ta pozostaje prawdziwa nawet dla proszków znacznie różniących się średnim rozmiarem ziarna (tabela 5.2).
| Próbka | ||||||||
|---|---|---|---|---|---|---|---|---|
| k6v | 022 | 4.08 | 0.0319672 | 0.7295257 | 141.9 | 119.7 | 1.18 | |
| k6v | 113 | 4.78 | 0.0333162 | 0.7295257 | 134.2 | 115.5 | 1.16 | |
| k6v | 222 | 4.99 | 0.0333162 | 0.7176996 | 109.9 | 94.4 | 1.16 | |
| k8 | 022 | 4.10 | 0.0164973 | 0.7443229 | 279.2 | 230.5 | 1.21 | |
| k8 | 044 | 8.18 | 0.0185272 | 0.75501 | 251.2 | 204.3 | 1.23 | |
| k8 | 113 | 4.79 | 0.0181521 | 0.7737516 | 260.7 | 206.8 | 1.26 | |
| 157k | 022 | 4.10 | 0.2026393 | 1.1512892 | 27.8 | 15.7 | 1.77 | |
| 157k | 113 | 4.78 | 0.2138783 | 1.1921648 | 26.6 | 14.6 | 1.82 | |
| ew3k | 022 | 4.10 | 0.2476773 | 1.3118399 | 23.5 | 12.1 | 1.95 | |
Po przeanalizowaniu wszystkich posiadanych materiałów istotnie dało się je pogrupować według właściwej im względnej szerokości rozkładu wielkości ziarna, rys. 5.14. Z rys. 5.14 widać, że zależnie od genezy proszków nanokrystalicznych, ich względne dyspersje różnią się nawet dwa razy: otrzymywany z eksplozji (II) ma rozkład wielkości krystalitów przeszło dwukrotnie szerszy niż (V) posiadający ziarna podobnej wielkości, lecz otrzymywany z izotermicznego wygrzewania materiału amorficznego (por. §5.1.1.1 i §5.1.1.2). Węglik krzemu syntezowany w płomieniu posiada wartość lokującą się pomiędzy tamtymi dwoma.
Zaobserwowano następujące ogólne prawidłowości:
-
•
proszki otrzymywane przez wzrost z fazy gazowej (synteza z węgla uwalnianego z materiału wybuchowego, płomień silanu) mają szersze rozkłady wielkości ziaren niż materiał syntezowany w ciele stałym (piroliza krzemoorganików, synteza diamentu z grafitu). Wiąże się to prawdopodobnie transportem masy w trakcie syntezy.
-
•
proszki otrzymywane w warunkach dużego gradientu temperatur, w silnie egzotermicznych, gwałtownych procesach w ośrodku o małej przewodności cieplnej (przemiana fazowa grafitu otaczającego jądro wybuchu, płomień silanu) mają szersze rozkłady wielkości ziaren niż materiał syntezowany w mniejszych gradientach temperatury i ośrodkach lepiej przewodzących ciepło. Wiąże się to prawdopodobnie transportem ciepła w trakcie syntezy.
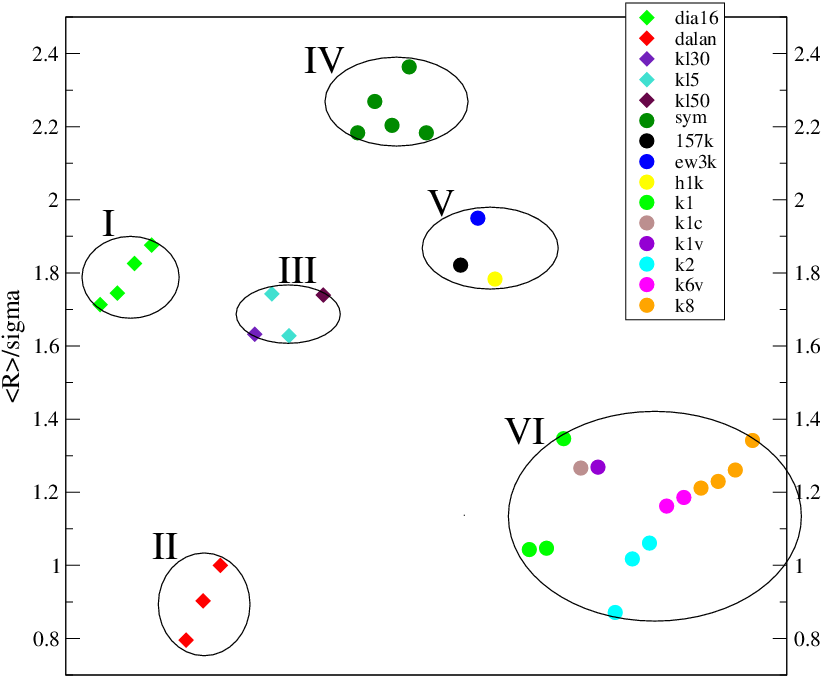
Można powiedzieć, że najwęższe rozkłady wielkości ziaren osiąga się w kwazistatycznych procesach wzrostu w ciele stałym (np. podczas długotrwałego wygrzewania materiału amorficznego w piecu o stałej temperaturze), zaś najszersze w syntezach gazowych przy dużym gradiencie temperatury.
5.3.3 Wnioski
Z doświadczalnych pomiarów szerokości rozkładu wielkości ziaren proszków otrzymywanych różnymi technikami wynika, że procesy transportu masy i ciepła w trakcie syntezy proszków nanokrystalicznych mają silny wpływ na rozrzut ich wielkości.
Spośród proszków otrzymywanych czterema technikami, najmniejszą względną szerokość zapewniła synteza w warunkach kwazistatycznych (grupa V, stała temperatura), rys. 5.14. Jest to proces prowadzony w amorficznym ciele stałym w warunkach izotermicznych, co zapewnia jednolite warunki wzrostu w całej objętości substratu. Uzyskaną względną szerokość rozkładu (dyspersja około dwukrotnie mniejsza od średniego rozmiaru ziarna) można więc traktować jako najmniejszą możliwą do uzyskania. Jest to minimum wynikające z samej termodynamiki procesu wzrostu (która nie jest tutaj dyskutowana) i wiele węższych rozkładów w tym mechanizmie wzrostu uzyskać się nie da.
Najszersze otrzymano dla proszków syntezowanych z węgla uwalnianego z materiału wybuchowego w czasie jego eksplozji (grupa II). Są one około dwukrotnie szersze niż poprzednie (dyspersja porównywalna lub nieco mniejsza od średniego rozmiaru ziarna). Możliwość powstawania proszków o jeszcze szerszych jest prawdopodobnie fizycznie ograniczona przez szybkość transportu ciepła i masy w gorącym gazie powstałym wskutek wybuchu. Ponieważ warunki w jądrze eksplozji są już ekstremalne (, ) nie należy się spodziewać dyspersji wiele większych niż wartość średniego rozmiaru ziarna.
Te dwa wnioski pokazują, że zakres dyspersji rozkładu wielkości ziaren możliwych do otrzymania w procesach wzrostu z fazy amorficznej jest skończony i jego oszacowanie można podać jako:
| (5.2) | |||||
Jeszcze raz spójrzmy na zależność stałej Scherrer’a od “monodyspersyjności” rozkładu wielkości ziaren (rys. 3.17). Przewidywane dla syntetyzowanych wszystkimi metodami wartości od do odpowiadają zakresowi silnej zmienności . Oznacza to, że duży błąd wyznaczanego z równania Scherrer’a rozmiaru ziarna w opisanym przypadku dotyczył wszystkich badanych nanoproszków. Ta sama uwaga odnosi się do wyników uzyskiwanych metodą Warren’a-Averbach’a (rys. 3.20): dyspersja rozmiarów ziaren w rzeczywistych proszkach jest zbyt duża, aby wyniki te mogły być dokładne.
5.4 Niskokątowe pomiary mikrostruktury nanoproszków
Ze względu na rozbudowaną powierzchnię małych ziaren i duże siły adhezji wiążące ziarna w trwałe struktury, nanoproszki nie są mieszaniną oddzielnych krystalitów, lecz podczas syntezy poszczególne ziarna łączą się w rozgałęzione łańcuchy, które luźno wypełniają przestrzeń, tworząc struktury fraktalne. Skłonność nanokryształów do tworzenia fraktali masowych jest funkcją ich rozmiaru: w proszkach o ziarnie poniżej takie zachowanie jest regułą. Wzrost fraktala odbywa się we wszystkich kierunkach: do zarodka dołączają nowo zsyntetyzowane krystality i tworzące się w ten sposób ziarno fraktalne może osiągać makroskopowe rozmiary, jak na rys. 5.15. Gęstość ziarna fraktalnego nie jest stała i maleje wraz z odległością od jego środka (zarodka):
| (5.3) |
gdzie jest masowym wymiarem fraktalnym zawsze mniejszym od . Ponieważ najczęściej , proszki nanokrystaliczne są niekiedy bardzo lekkie (kilkanaście gramów wypełnia objętość ). Dzięki związkowi (5.3) gęstości ziaren fraktalnych z wymiarem , rozpraszanie niskokątowe promieni rentgenowskich (Small Angle X-Ray Scattering, ), które odbywa się na zmianach gęstości elektronowej, umożliwia wyznaczanie wymiaru . Chociaż fraktalne ziarna nanokrystalicznych i są dość odporne mechanicznie, przyłożenie dużego nacisku podczas zagęszczania proszku łamie gałęzie fraktala i zmienia mikrostrukturę proszku.
W tym rozdziale przedyskutowany zostanie wpływ ciśnienia na mikrostrukturę nanoproszków i zbadany przy pomocy rozpraszania niskokątowego.

5.4.1 Materiał, eksperyment i analiza danych
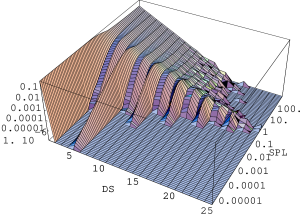
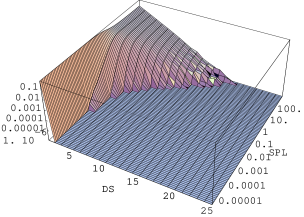
Do pomiarów rentgenowskiego rozpraszania niskokątowego () i ultraniskokątowego () użyto sześciu zestawów próbek nanokrystalicznych i . Każdy z -ciu zestawów składał się z sześciu próbek tego samego materiału zagęszczonych w prasie izostatycznej w ciśnieniach , , , , i . Pierwszych pięć ciśnień otrzymano w prasie ręcznej, najwyższe - w prasie hydraulicznej. Pomiary przeprowadzono na dwóch liniach synchrotronu DESY w Hamburgu. Na linii B1 (rozpraszanie niskokątowe, por. §5.1.4.2) zmierzono krzywe w zakresie Å. Linia ultraniskokątowa BW4 umożliwiła rozszerzenie zakresu mierzonego rozpraszania na Å. Na obu stacjach badawczych zainstalowane są dwuwymiarowe detektory drutowe o rozdzielczości punktów i układ do prowadzenia wiązki rozproszonej w próżni. Otrzymany z detektora dwuwymiarowy profil niskokątowy (rys. 5.16) został zintegrowany do jednowymiarowej krzywej.
a)
|
b)
|
5.4.1.1 Wyznaczanie średniego rozmiaru kryształów
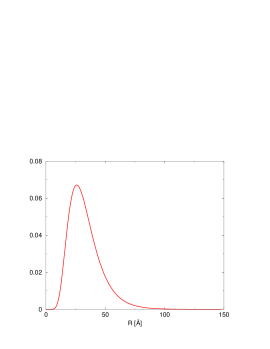
Średnią wielkość drobin (krystalitów) określa się z profilu niskokątowego używając przybliżenia Guinier [13, str.126]:
| (5.4) |
gdzie jest analizowaną krzywą niskokątową. Po wykreśleniu jej jako logarytmu dziesiętnego natężenia w funkcji kwadratu wektora rozpraszania (tzw. krzywa Guinier, rys. 5.17b), średni rozmiar drobin otrzymujemy ze związku:
| (5.5) |
gdzie jest zmierzonym nachyleniem krzywej Guinier w pobliżu wartości wektora rozpraszania spełniających warunek , rys. 5.17a. Obszar jest łatwy do rozpoznania na krzywej wykreślonej w skali log-log, gdyż ogranicza od lewej zakres , gdzie spełnione jest prawo Porod’a (), por. §3.4.3. Na rys. 5.17a obszar ten zaznaczono elipsą.
a)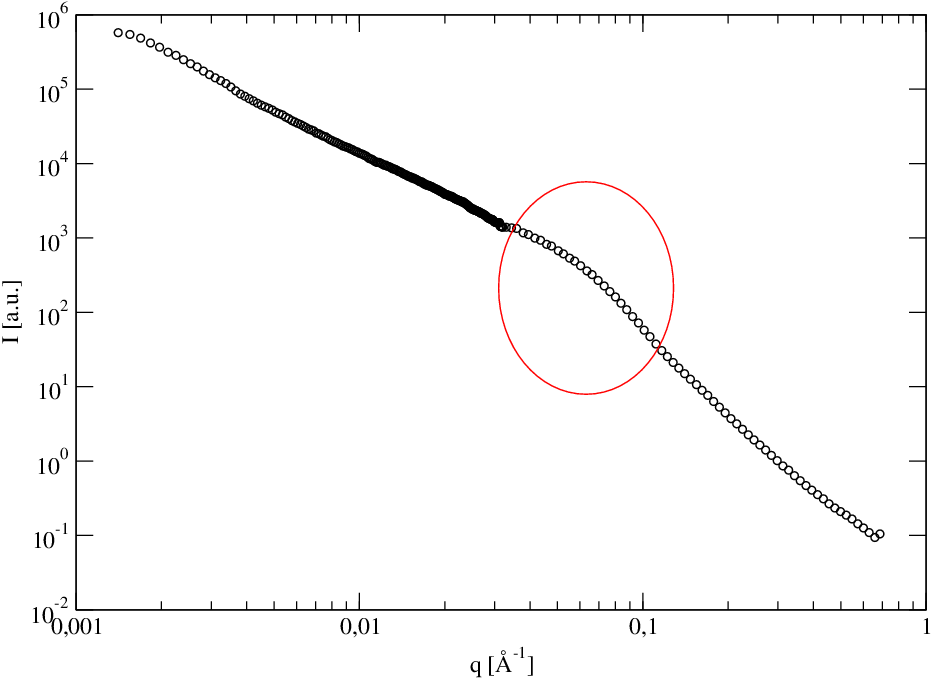
|
b)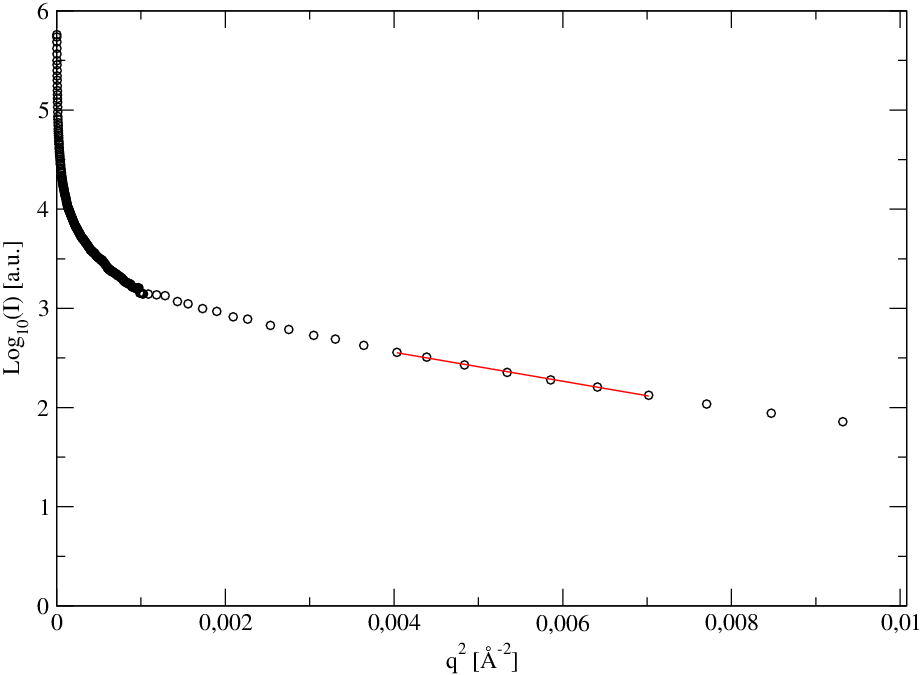
|
5.4.1.2 Charakteryzacja aglomeratów i struktury fraktalnej proszku
Szczególną cechą rozpraszania niskokątowego jest - obok pomiarów wielkości ziaren - możliwość badania obiektów większych niż rozmiar ziarna proszku (lewa część krzywej z rys. 5.17a). Obiekty te to układy ziaren pozostające w pewnych geometrycznych relacjach względem siebie. Spodziewane relacje to:
-
1.
Brak jakichkolwiek relacji. Luźny proszek z niepowiązanymi, stosunkowo odległymi od siebie ziarnami, najlepiej zawiesina drobin w cieczy. Krzywa takiego układu dla powinna mieć stałe nachylenie .
-
2.
Aglomeraty ziaren. Część ziaren związana w “super ziarna” o wielokrotnie większym rozmiarze niż ziarna pierwotne (krystality). Krzywa powinna składać się z segmentów o nachyleniu właściwym dla każdego “podproszku”, występujących w obszarze właściwym dla średniego rozmiaru ziarna (lub aglomeratu) każdego “podproszku”.
-
3.
Fraktale masowe. Rozgałęzione aglomeraty ziaren mogące osiągać rozmiary porównywalne z objętością próbki (rys. 5.15). Rozpraszają jak pojedyncze ziarna ale o zmiennej gęstości. Gęstość fraktala masowego o wymiarze jest funkcją odległości od jego centrum: . Rozpraszanie na takich strukturach daje, według [48], nachylenia krzywej niskokątowej .
Krzywa niskokątowa posiada typowo trzy rejony niosące trzy różne informacje:
-
•
część wysokokątową o - zazwyczaj - stałym nachyleniu właściwym dla formy rozkładu wielkości ziaren proszku (por. §3.5.7),
- •
-
•
część ultraniskokątową, której nachylenie pozwala określić masowy wymiar fraktalny struktury tworzonej przez ziarna proszku i/lub aglomerację ziaren.
5.4.2 Wyniki
Wyniki pomiaru nachyleń pierwszej części krzywej (rozkład wielkości ziaren) podano w tabeli 5.3. Badane przez nas nanometrowe proszki i niezależnie od ciśnienia, w którym zagęszczano próbkę wykazują zawsze podobne nachylenie bliskie . Świadczy to o niezmienności postaci rozkładu wielkości krystalitów w ciśnieniu, co jest wynikiem oczekiwanym.
| Próbka \ Ciśnienie | ||||||
|---|---|---|---|---|---|---|
| 157k | ||||||
| ew3k | ||||||
| k9 | ||||||
| 493-158-3 | ||||||
| a16 | ||||||
| atm | ||||||
Wyniki pomiaru średniego rozmiaru ziarna metodą Guinier (zagięcie krzywej ) dla wszystkich badanych próbek zestawiono w tabeli 5.4.
| Próbka | ||||
|---|---|---|---|---|
| 157k | 146.12 | 32Å | 27Å | |
| ew3k | 141.69 | 31Å | 24Å | |
| k9 | 1819.7 | 112Å | - | |
| 493-158-3 | 1701.1 | 108Å | - | |
| a16 | 135.9 | 31Å | - | |
| atm | 123.71 | 29Å | - | |
W przypadku dwóch próbek (157k i ew3k), których mikrostrukturę badano niezależnie na podstawie profilu linii bragowskich w §5.3, podano odpowiednie wartości średniego rozmiaru krystalitów . Istnieje systematyczna zależność między rozmiarami wyznaczonymi z maksimów niskokątowych i bragowskich: pierwsze z nich są zawsze nieco większe.
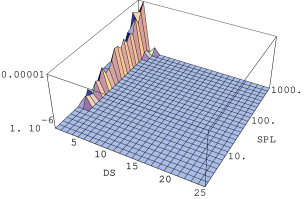
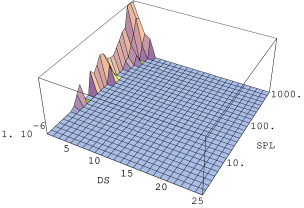
Nachylenie części ultraniskokątowej krzywej informuje m.in. o wymiarze fraktalnym struktury tworzonej przez ziarna proszku. Nachylenia tej części w zależności od ciśnienia użytego podczas przygotowania próbek podano w tabeli 5.5. Pięć na sześć badanych próbek posiadało fraktalną strukturę ziaren o wymiarze pomiędzy i . Jedynie proszek o największych ziarnach (k9, Å) ma wymiar mniejszy od jedności, co oznacza krótkie, jednowymiarowe ciągi pozlepianych ziaren. Widać wyraźną zależność wymiaru fraktalnego od ciśnienia przyłożonego podczas formowania próbek.
| Próbka \ Ciśnienie | - | ||||||
|---|---|---|---|---|---|---|---|
| 157k | |||||||
| ew3k | |||||||
| k9 | |||||||
| 493-158-3 | |||||||
| a16 | |||||||
| atm | |||||||
a)
b)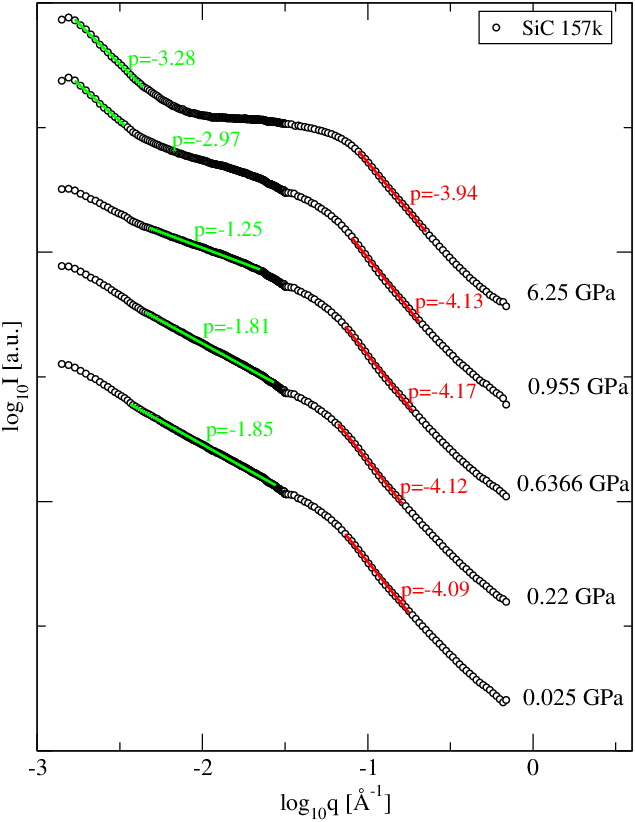
|
c) 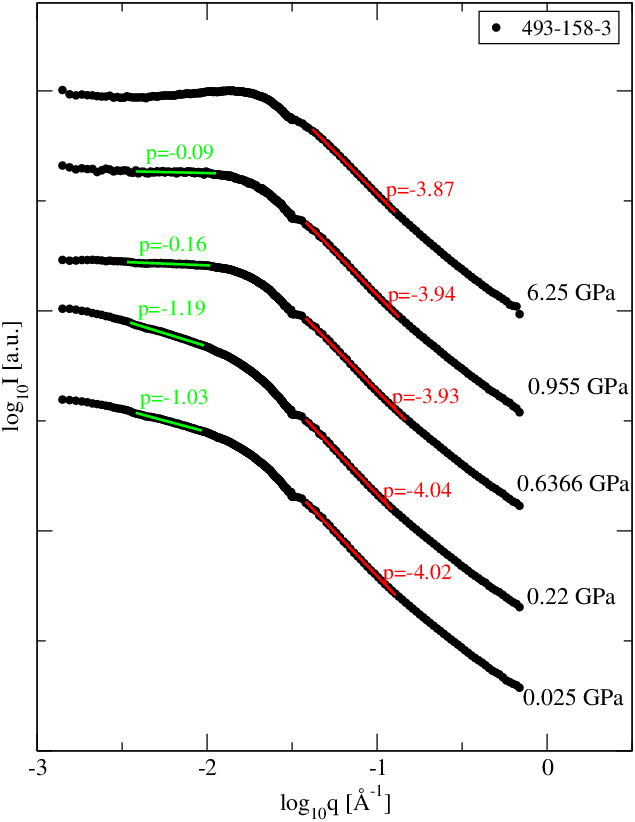
|
5.4.3 Dyskusja wyników i wnioski
5.4.3.1 Pomiar rozmiaru ziarna i zanieczyszczeń powierzchni
Porównanie wartości średniego rozmiaru ziarna zmierzonego w dwóch eksperymentach: (i) dyfrakcji bragowskiej i (ii) rozpraszania niskokątowego przeprowadzonych dla tego samego proszku może dostarczyć informacji na temat substancji zaadsorbowanych na powierzchni nanokryształów. Jest to możliwe, gdyż oba doświadczenia wyznaczają rozmiar ziarna bazując na nieco innych mechanizmach fizycznych:
-
•
Wielkość ziarna wyznaczana z profilu linii bragowskich ma znaczenie rozmiaru koherentnie rozpraszającej struktury krystalicznej. Wkłady rozproszeń pochodzące od atomów związanych na powierzchni nanokryształu ale nie znajdujących w węzłach jego sieci krystalicznej znoszą się wzajemnie nie uczestnicząc w konstruktywnej interferencji. Nie stanowią one składnika bragowskich maksimów interferencyjnych, dlatego pomiar szerokości tych maksimów mówi o rozmiarze nanokryształu “netto”, bez otoczki obcych molekuł związanych na jego powierzchni.
-
•
Rozpraszanie niskokątowe odbywa się na różnicach gęstości elektronowej, w proszkach - na granicach ziaren66 6 Oczywiście w rzeczywistości promieniowanie podlega jednakowym prawom niezależnie od kąta i zawsze rozprasza się na elektronach w całej objętości próbki. Uzasadnienie uproszczenia można znaleźć w równaniu Debye’a: małe odległości międzyatomowe (wewnątrz jednego ziarna) wnoszą w obraz dyfrakcyjny wolnozmienne w obszarze niskokątowym składowe i profil efektywnie formują składowe pochodzące od dużych odległości międzyziarnowych (szybkozmienne w obszarze niskich kątów).. Dla niskokątowego maksimum padające promieniowanie jest przez atomy “uginane w przód”, a tym samym fazy fal rozproszonych na wszystkich atomach automatycznie spełniają warunek konstruktywnej interferencji. I to niezależnie od pozycji atomu (w sieci krystalicznej lub poza nią). Dlatego nie jest czułe na strukturę atomową materiału i otrzymywany tą techniką rozmiar ziarna jest rozmiarem “brutto”, czyli uwzględnia krystalit łącznie ze wszystkimi zaadsorbowanymi na jego powierzchni molekułami.
Zgodnie z powyższym, różnica rozmiarów wyznaczanych dla tych samych proszków z linii bragowskich i z krzywych powinna odpowiadać grubości otoczki nanokryształu złożonej z zaadsorbowanych na nim obcych atomów (rys. 5.19a). Trzeba jednak wyraźnie powiedzieć, że do zaobserwowania tak nieznacznych różnic rozmiaru konieczna jest niezwykle ostrożna analiza danych doświadczalnych. Np. sama obecność błędów ułożenia powoduje poszerzenie linii odpowiadające znacznie większym różnicom rozmiarów niż spodziewana grubość zanieczyszczeń okołokrystalicznych. Dlatego autor odważył się porównywać rozmiary ziaren otrzymanych z danych niskokątowych wyłącznie z rozmiarami pochodzącymi z metody ab initio wyznaczania rozkładu wielkości ziaren (metoda ta uwzględnia obecność błędów ułożenia w strukturze, por. §5.2).
Na podstawie systematycznej różnicy można przypuszczać, że badane próbki nanokrystalicznego posiadały na swojej powierzchni warstwę obcych molekuł o grubości ok. Å, tab. 5.4. Prosty eksperyment przeprowadzony w czasie przygotowywania próbek nanokrystalicznego do pomiarów dyfrakcyjnych pozwala na stwierdzenie, że na powierzchni ziaren adsorbuje się powietrze: nanoproszek umieszczony w eksykatorze próżniowym podczas wypompowywania zeń powietrza gwałtownie “wrze” przez kilkanaście sekund. Po doprowadzeniu powietrza na kilka minut i ponownym jego odpompowaniu wrzenie następuje znów, ale jest słabsze. Rzeczywiście, promień kowalencyjny azotu i tlenu wynosi ok. Å, a więc całkowita długość cząsteczki i wynosi ok. Å. Odpowiada to zmierzonej dyfrakcyjnie grubości otoczki molekularnej na nanokryształach , rys. 5.19a. Hipotezę o zaadsorbowanym na nanokryształach powietrzu potwierdzają także pomiary (Thermal Gravity Analysis) w połączeniu ze spektrometrią masową: nanokryształy po wygrzaniu w próżni stają się lżejsze o masę odpowiadającą w przybliżeniu pojedynczej warstwie cząsteczek gazu na swej powierzchni (tlen i azot mają zbliżone masy cząsteczkowe). Obok azotu i tlenu emitowane są , , , i . Na rysunku 5.19b pokazano spadek ubytek masy nanoproszku w trakcie jego wygrzewania w strumieniu gazów szlachetnych: argonu i helu, unoszących z materiału uwolnione zanieczyszczenia.
a) 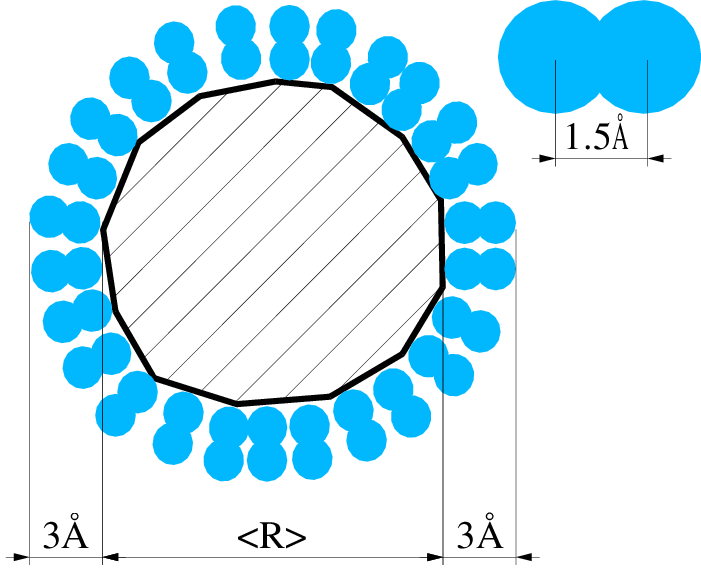
|
b)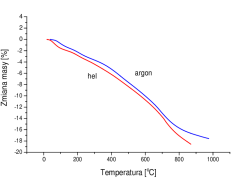
|
5.4.3.2 Przemiany mikrostruktury proszku w czasie wysokociśnieniowego zagęszczania
Proces zagęszczania nanoproszków polikrystalicznych, można podzielić na pięć etapów [36]:
-
1.
Wyjściowy materiał składa się z mieszaniny pojedynczych ziaren, ich łańcuchów i aglomeratów. Stopień zagęszczenia jest mały i proces przebiega dzięki elastyczności wiązań pomiędzy ziarnami. Na tym etapie wyjściowa sieć ziaren odkształca się elastycznie ale ziarna nie przemieszczają się względem siebie.
-
2.
Zagęszczanie przyspiesza z powodu odkształceń kwazi-plastycznych. Sieć ziaren deformuje się i przebudowuje poprzez poślizgi i obroty ziaren. Przemieszczanie ziaren w nanoproszkach jest ograniczone dużymi siłami tarcia ziaren. Opory spowodowane tarciem są znaczne ze względu na liczne punkty kontaktów między ziarnami.
To zachowanie może być porównane do przekroczenia granicy plastyczności w ciałach stałych. Należy jednak zaznaczyć, że nie jest ono powodowane własnościami sieci krystalicznej czy jej defektów lecz własnościami powierzchni i granic ziaren. -
3.
Sieć ziaren odkształca się elastycznie i plastycznie do momentu, kiedy nacisk osiąga wartość krytyczną - “granicę płynięcia”. W tych warunkach ciśnienia pękają wiązania pomiędzy poszczególnymi ziarnami. Składowa ścinająca przyłożonego nacisku powoduje przemieszczenia ziaren i zapadanie dużych porów. Przebudowa struktury ziaren i makroskopowa deformacja porów zwiększa liczbę kontaktów między ziarnami. Stykające się ziarna obracają się i ślizgają aby osiągnąć minimum energii powierzchniowej.
-
4.
Proszek stale zagęszcza się w miarę postępu kwazi-plastycznej przebudowy układu ziaren. Zagęszczanie przebiega względnie szybko dopóki materiał nie osiągnie gęstości bliskiej wartości teoretycznej.
-
5.
Gęsty i jednolity kompakt nanokrystaliczny jest ściskany dalej tak jak na to pozwalają własności mechaniczne jego sieci krystalicznej i granic ziaren. Stanowi on na tym etapie jednolity materiał.
Pierwszy etap, który nie powoduje jeszcze przemieszczania ziaren a jedynie naprężenia ich sieci krystalicznej następuje dla badanych nanoproszków i w ciśnieniach ok. (tab. 5.5 i rys. 5.18b i c). W ciśnieniu obserwuje się pierwsze istotne zmiany struktury fraktalnej ziaren - jest to drugi etap zagęszczania. We wszystkich badanych proszkach (tab. 5.5) stwierdzono zmniejszenie masowego wymiary fraktalnego , co jest spowodowane załamywaniem się w tym ciśnieniu włókien pozrastanych nanokryształów. Proszek gwałtownie zmniejsza swoją objętość. Trzeci etap zagęszczania zaobserwowano w ciśnieniu ok. . Zakończony zostaje proces degradacji struktury fraktalnej. Proszek składa się bądź z osobnych ziaren bądź z bardzo krótkich ich łańcuchów, które przemieszczają się względem siebie szukając orientacji minimalizującej energię powierzchniową. Dla jednej z próbek (rys. 5.18c) zaobserwowano niewielkie maksimum interferencyjne wskazujące na tworzenie się periodycznej sieci ziaren w proszku. W czterech z sześciu próbek, w najwyższym ciśnieniu , nachylenie ultraniskokątowej części krzywej było większe niż w minimalnym ciśnieniu (tab. 5.5). Jest to spowodowane tworzeniem się z rozbitych fraktali gęstych aglomeratów ziaren o rozmiarach kilkuset . Czwarty i piąty etap zagęszczania nie zostały osiągnięte dla przyłożonych w eksperymencie ciśnień.
Fraktalność struktury proszków nanokrystalicznych jest faktem ważnym ze względu na zastosowania. Wytrzymała mechanicznie (nie degraduje się do atmosfer!) sieć obiektów o rozmiarach kwantowych pozwalająca związać (i uwolnić) na swojej powierzchni tyle gazu ile sama waży77 7 Dla ziaren o średnicy Å. (czyli około na każdy gram nanoproszku) posiada wiele potencjalnych zastosowań. Jednym z nich jest bezpieczne, niskociśnieniowe przechowywanie materiałów pędnych (np. wodoru). Ważna jest przy tym nie tylko powierzchnia aktywna ale i sama budowa fraktala (gęstość malejąca od centrum ziarna), która pozwala na szybkie uwalnianie dużego strumienia cząstek. Innym przykładem zastosowań fraktalnych ziaren nanometrowych jest optoelektronika. Świecący na niebiesko porowaty krzem z wodorem związanym na swojej powierzchni wywołał swego czasu duże poruszenie wśród fizyków szukających źródeł niebieskiego światła. Spiekanie z nanoproszków gęstych, supertwardych i plastycznych ceramik wymaga ich zagęszczania, a więc znajomości mechanizmu i warunków w jakich fraktale nanokryształów się degradują [8][38][45][34][35][44][40][9]. Pokazano, że następuje to w izostatycznym ciśnieniu rzędu kilku .
5.5 Wpływ ciśnienia na strukturę nanokryształów
5.5.1 Wprowadzenie
Relaksacja naprężeń sieci krystalicznej w nanometrowych polikryształach azotku galu jest procesem, który jednocześnie zmienia strukturę i mikrostrukturę tego materiału. W odróżnieniu od materiałów supertwardych, takich jak węglik krzemu czy diament (por. [43][39]), nanokryształy ulegają odkształceniom plastycznym wobec stosunkowo niewielkich88 8 Nanoproszek ściskany w warunkach niehydrostatycznych ulega odkształceniom plastycznym w ciśnieniach rzędu pojedynczych podczas gdy odkształcenia nanokryształów i są wyłącznie elastyczne przynajmniej do ciśnień rzędu (tyle wynosi największe uzyskane przez nas ciśnienie). sił przykładanych do ich ścian. Naprężenia sieci krystalicznej relaksują wtedy poprzez ruch dyslokacji, początkowo wzdłuż płaszczyzn , gdyż te mają w strukturach hexagonalnych najmniejszą energię aktywacji. Przemieszczenia płaszczyzn krystalicznych w tym kierunku zmieniają sekwencję politypową krystalitu (a więc jego strukturę) co stanowi mierzalny metodami dyfrakcyjnymi ślad po całym procesie. Poniżej prezentujemy wyniki symulacji numerycznej wprowadzania błędów ułożenia do struktury . Dyfraktogramy obliczone metodą ab initio na poszczególnych jego etapach zostaną porównane do danych doświadczalnych zmierzonych dla we wzrastających ciśnieniach niehydrostatycznych (por. [41][12][11] oraz [37]).
5.5.2 Materiał i eksperyment
Zbadano nanokrystaliczny proszek o ziarnach wielkości zsyntezowany z polimerów metaloorganicznych [18]. Nanokryształy zostały poddane działaniu wysokiego ciśnienia, do , w komórce diamentowej (Diamond Anvil Cell, ) w temperaturze pokojowej, rys.5.9. Dane dyfrakcyjne zebrano w geometrii z ustalonym kątem detektora czułego na fotony o różnych energiach (energy dispersive), por. paragraf 5.1.4.1. Zewnętrzny nacisk był przenoszony na nanokryształy bez pośrednictwa ośrodka ciśnieniowego (warunki niehydrostatyczne). Pomiar ciśnienia odbywał się poprzez badanie parametru sieci złota, dodanego w niewielkiej ilości do próbek .
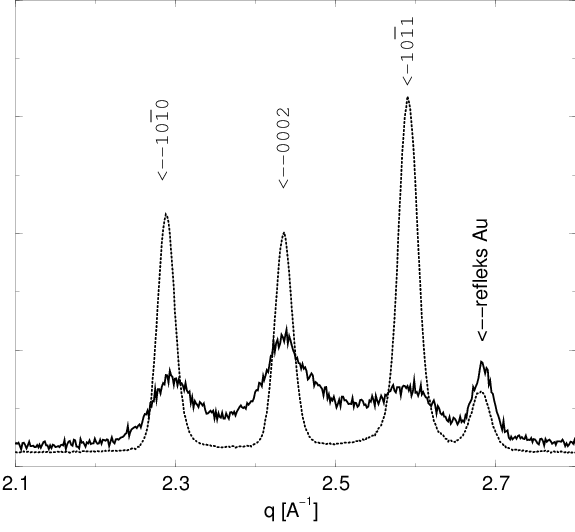
Zaobserwowano zmianę względnych natężeń linii dyfrakcyjnych połączoną z ich silną deformacją i podniesieniem poziomu tła, rys. 5.20. Wskazuje to na strukturalne przemiany w azotku galu w warunkach silnych naprężeń.
5.5.3 Model wprowadzania błędów ułożenia
Błędy ułożenia mogą być wprowadzane do kryształów o strukturze najgęstszego upakowania podczas ich wzrostu bądź wskutek relaksacji naprężeń sieci krystalicznej. Podczas wzrostu nowych warstw kryształu nie ma ograniczeń co do wyboru pozycji politypowych tych warstw, dlatego tworząca się struktura błędów ułożenia może być dowolna (w szczególności możliwe są błędy typu extrinsic, por. 5.1.2). Inaczej jest w przypadku istniejącego już kryształu z naprężeniami sieci: dla ich rozładowania musi nastąpić jej częściowa rekonstrukcja (odkształcenie plastyczne). Jednak nie każdy mechanizm dyslokacyjny, który rekonstrukcję umożliwia jest jednakowo prawdopodobny. W strukturach najgęstszego upakowania, szczególnie zawierających błędy ułożenia, najbardziej prawdopodobna płaszczyzna poślizgu to [22, str.587]. Dlatego w warunkach stopniowo wzrastającego nacisku (a więc i naprężeń) w pierwszym rzędzie można oczekiwać poślizgów wzdłuż tych właśnie płaszczyzn (w coraz wyższych ciśnieniach uruchamiać się będą również mechanizmy dyslokacyjne dla płaszczyzn krystalicznych o większych energiach aktywacji ruchu dyslokacji). Poślizgi wzdłuż płaszczyzn oprócz niskiej energii aktywacji mają też tę szczególną cechę, że przemieszczenie (czyli wektor poślizgu) nie musi być równy wektorowi translacyjnemu sieci [22, str.588]. Częściowe przemieszczenie zmienia strukturę politypową kryształu i daje tym samym szansę obserwacji postępów procesu relaksacji naprężeń metodami dyfrakcyjnymi. Wykorzystując prosty model przesunięć warstw i opisane w paragrafie 4.2.1 obliczenia ab initio dyfrakcji na nanokryształach postaramy się prześledzić proces powstawania błędów ułożenia spowodowany naprężeniami sieci krystalicznej w czystej strukturze na przykładzie nanokryształów azotku galu.
Aby wyjaśnić obserwowane doświadczalnie zmiany dyfraktogramów przyjęty został prosty model powstawania błędów ułożenia w wysokim ciśnieniu niehydrostatycznym. W pierwszej fazie zagęszczania proszku nanokrystalicznego ziarna przemieszczają się względem siebie formując układ zbliżony do najgęściej upakowanego. W tej fazie mamy do czynienia z łamaniem ewentualnych struktur fraktalnych ziaren i gwałtownym zmniejszaniem objętości próbki. Po zakończeniu tego procesu ziarna proszku zostają “uwięzione” w swoich położeniach przez swych sąsiadów. Dalsze zwiększanie nacisku nie powoduje już gwałtownego zapadania się materiału - pojawiają się natomiast silne naprężenia. Wobec braku ośrodka ciśnieniowego nanokryształy oddziaływają ze sobą bezpośrednio. Naprężenia powstające w punktach styku ziaren relaksują poprzez poślizgi wzdłuż płaszczyzn . Obserwuje się, że deformacja kryształu przez poślizg jest niejednorodna [22]. Mamy do czynienia z jednoczesnymi poślizgami wzdłuż wielu równoległych płaszczyzn kryształu, pomiędzy którymi istnieje niezdeformowana jego część. W nanokryształach płaszczyzny te są siłą rzeczy nieodległe i w granicznym przypadku cały proces deformacji plastycznej można przedstawić jako ciąg przesunięć pojedynczych warstw przy zachowaniu niezmienionego położenia wszystkich atomów nienależących do aktualnie przesuwanej warstwy.
Proces przesuwania pojedynczych płaszczyzn prowadzi do zmian sekwencji politypowej w krysztale, rys. 5.21.
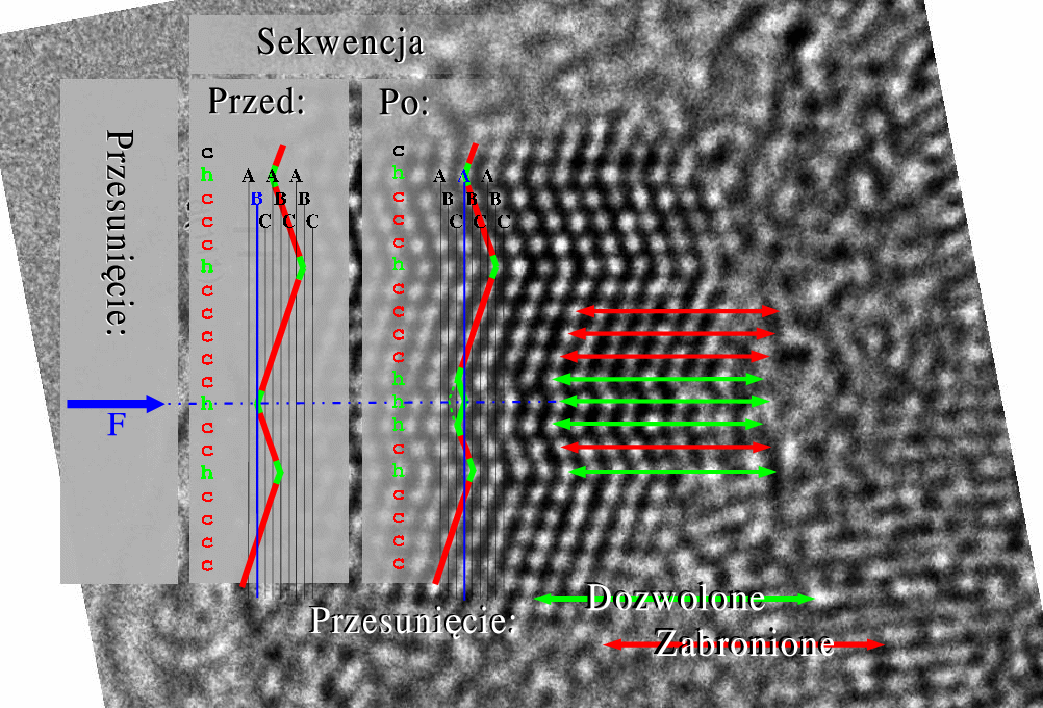
Jednak nie wszystkie sekwencje politypowe mogące się pojawić w opisywanym procesie są dozwolone: dwie sąsiednie płaszczyzny nie mogą być w tych samych pozycjach (…AA…, …BB…, …CC…). Z racji tych ograniczeń uwzględnione mogą być tylko przesunięcia nie prowadzące do zabronionych konfiguracji warstw. Jak łatwo stwierdzić tylko czysta struktura hexagonalna () daje całkowitą swobodę wyboru miejsca pierwszego przesunięcia: każda z warstw ma wtedy obie sąsiednie warstwy będące w tej samej pozycji (np. …ABABABABAB…: każda warstwa B jest otoczona przez dwie A i odwrotnie, patrz paragraf 5.1.1), dzięki czemu sama może przejść do alternatywnej pozycji (np. z B do C lub z A do C). Po wprowadzeniu pierwszego i następnych błędów ułożenia rośnie liczba warstw kubicznych w związku z czym w procesie relaksacji naprężeń zwiększa się ilość przesunięć niedozwolonych. Proces ten, prowadzony w opisany sposób, zbiega do struktury całkowicie nieuporządkowanej, rys. 5.22. Struktura nieuporządkowana składa się z mieszaniny domen typu i o różnej długości, (por. paragraf 5.1.2.2).
Przeciwnie do czystej struktury , czysta struktura gwarantuje niemal całkowitą odporność na przesunięcia wprowadzane według proponowanego modelu. W strukturze kubicznej prawie każda warstwa sąsiaduje z dwiema innymi, będącymi w różnych pozycjach (np. ABCABCABC: A sąsiaduje z B i C, B sąsiaduje A i C, C sąsiaduje z A i B), co zmusza ją do pozostawania w pozycji wyjściowej, gdyż dowolne jej przesunięcie powodowałaby powstanie zabronionej sekwencji dwóch identycznych warstw. “Prawie” każda, gdyż dwie warstwy powierzchniowe mają tylko jednego sąsiada, a co za tym idzie - możliwość relaksacji naprężenia przez przesunięcie do innej pozycji. Powierzchnia jest jedynym miejscem, gdzie mogą tworzyć się błędy ułożenia. Pomimo tego, że w przypadku kryształów nanometrowych dwie warstwy powierzchniowe stanowią istotną część wszystkich warstw kryształu, rozporządkowanie struktur poprzez przesunięcia pojedynczych warstw jest zdecydowanie trudniejsze niż jakichkolwiek innych politypów, rys.5.23.


Symulację wprowadzania jednowymiarowego nieporządku prowadzono według następującego algorytmu:
-
1.
Ustal wyjściową sekwencję politypową
np. 19-warstwowa sekwencja ’BA BCAB ACBACB CAB ACBA’, jak na rys. 5.21 -
2.
Wybierz losowo warstwę - kandydata na przesunięcie
np. 12-tą warstwę sekwencji ’BA BCAB ACBACB CAB ACBA’, z rys. 5.21 -
3.
Sprawdź, czy jej przesunięcie nie doprowadzi do niefizycznej sekwencji
jeśli nie: kontynuuj
jeśli tak: wróć do kroku 2 (ponów losowanie)
tutaj: przesunięcie prowadzi do sekwencji ’BA BCAB ACBACA CAB ACBA’, dozwolonej, jak na rys. 5.21 -
4.
Dokonaj przesunięcia warstwy (bieżąca sekwencja zmienia się)
nowa sekwencja jest teraz ’BA BCAB ACBACA CAB ACBA’, jak na rys. 5.21 -
5.
Kontynuuj od kroku 2 z bieżącą sekwencją
Prawdopodobieństwo udanego (fizycznego) przesunięcia warstwy w kroku 3-4 jest w zasadzie proporcjonalne do liczby warstw w krysztale. Liczba przesunięć określa stopień zaawansowania procesu rozporządkowywania struktury wyjściowej i jest unormowana do liczby warstw (Przesunięcia na warstwę, Shifts Per Layer, ). Określa ona liczbę wszystkich przesunięć, czy raczej “prób przesunięć”, włączając w to również te nieefektywne.
Proces implementacji błędów ułożenia według założonego mechanizmu można podzielić na dwie fazy, rys. 5.24b:
-
1.
wzrost hexagonalności w przedziale
-
2.
spadek hexagonalności powyżej
a)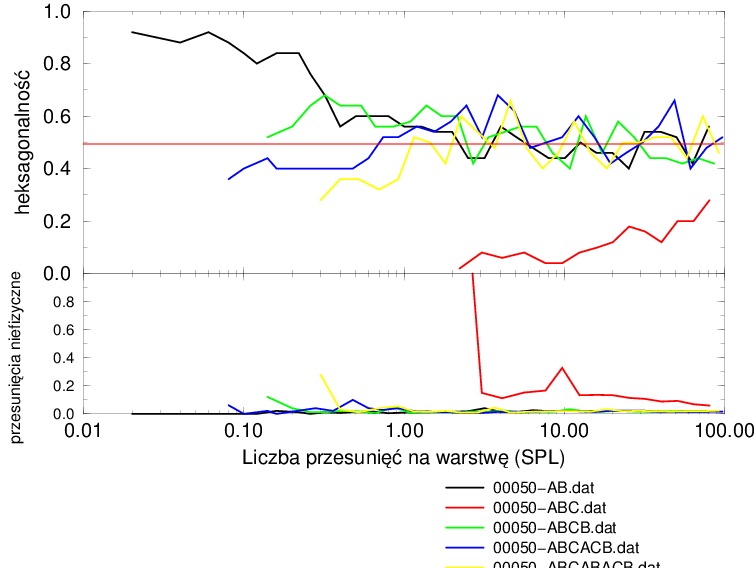
|
b)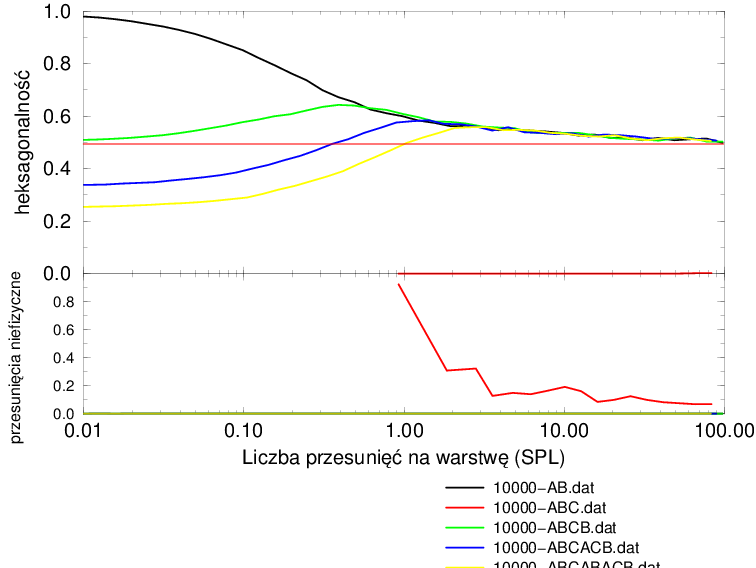
|
Aby wyjaśnić powody istnienia maksimum hexagonalności dla większości politypów potrzebna jest dokładniejsza analiza zmian sekwencji politypowych w trakcie procesu wprowadzania błędów ułożenia.
5.5.4 Operatory zmiany sekwencji
Zgodnie z przyjętym mechanizmem powstawania błędów ułożenia, efekt przesunięcia pojedynczej warstwy zależy od pozycji obu warstw z nią sąsiadujących. W tabeli 5.6 wymieniamy wszystkie warianty sekwencji politypowych mogących powstać na skutek przesunięcia pojedynczej warstwy dla trzech konfiguracji jej otoczenia:
-
•
przesuwamy samotną warstwę (’h’ otoczone warstwami typu ):
wynik: chchhh, kreacja dwóch dodatkowych warstw -
•
przesuwamy parę warstw typu (’hh’ otoczone warstwami typu ):
wynik: chhhhc, liczba warstw niezmieniona lecz wynikowa para jest przesunięta o jedną pozycję -
•
przesuwamy trójkę warstw typu (’hhh’ otoczone warstwami typu ):
wynik: chhhhhch, lub: hhhchchh, lub: hhhchc, wtrącenie warstwy w sekwencję bądź anihilacja dwóch warstw , zależnie od miejsca przyłożonego przesunięcia’h’ ’hh’ ’hhh’ I. wyjściowa sekwencja xcchccx xcchhccx xcchhhccx wyjściowa sekwencja ABCACBA ABCACABC ABCACACBA miejsce przesunięcia | | | nowa sekwencja ABCBCBA ABCBCABC ABCBCACBA nowa sekwencja xchhhcx xchhcccx xchhchccx wynik hhhh hhhh hhhhh+h II. wyjściowa sekwencja xcchhccx xcchhhccx wyjściowa sekwencja ABCACABC ABCACACBA miejsce przesunięcia | | nowa sekwencja ABCABABC ABCABACBA nowa sekwencja xccchhcx xccchcccx wynik hhhh hhhh III. wyjściowa sekwencja xcchhhccx wyjściowa sekwencja ABCACACBA miejsce przesunięcia | nowa sekwencja ABCACBCBA nowa sekwencja xcchchhcx wynik hhhhh+h Table 5.6: Możliwe warianty zmian sekwencji politypu spowodowane pojedynczym przesunięciem płaszczyzny dla trzech wyjściowych sekwencji: ’h’, ’hh’ i ’hhh’.
Łatwo zauważyć, że trzeci wynik jest złożeniem dwóch pierwszych, mianowicie:
-
•
przejście: hhhchc jest przejściem odwrotnym w stosunku do wyniku pierwszego
-
•
przejście: chhhhhch można rozłożyć na: przesunięcie chhhhc (wynik drugi) i zachowanie skrajnej warstwy niezmienionej hh
Analogiczna analiza coraz dłuższych sekwencji warstw typu (patrz np. rys. 5.22a) pokazałaby, że każdą przemianę sekwencji politypowej kryształu powstałą w oparciu o prezentowany model przesunięć płaszczyzn można zapisać jako złożenie dwóch podstawowych operacji:
| (5.6) | |||||
| (5.7) |
oraz odpowiednich operacji odwrotnych:
| (5.8) | |||||
| (5.9) |
5.5.5 Rozkład długości domen
Ewolucję rozkładu długości domen najdogodniej jest prześledzić na przykładzie bardzo długiej sekwencji czystego politypu , rys. 5.25.
-
•
W początkowej fazie procesu operator dzieli każdą długą wyjściową domenę na dwie mniejsze (wciąż duże), tworząc jedną izolowaną warstwę (wzrost liczebności domen o długości ) i dwie warstwy .
-
•
W pewnym momencie gwałtownie wzrasta prawdopodobieństwo znalezienia krótkiej99 9 Krótkiej w porównaniu do długości domeny wyjściowej, tj. długości rzędu kilkudziesięciu warstw. domeny hexagonalnej i to praktycznie niezależnie od jej długości. Nieco bardziej prawdopodobne są domeny najkrótsze1010 10 Zwłaszcza w przypadku nanokryształów; dokładniejsza dyskusja dalej.. Kryształ osiąga przejściowy stan nieuporządkowania, w którym rozkład długości domen jest “biały” (prawdopodobieństwo znalezienia każdej długości jest podobne).
-
•
W dalszym ciągu następuje rozbicie domen o bardzo zróżnicowanej długości na krótsze, o podobnych długościach. Jednocześnie operator tworzy przy każdym podziale domeny jedną izolowaną warstwę , ich liczba jest więc niepomiernie większa od domen o większych długościach, rys. 5.25b. Przy tej okazji anomalnie wzrasta też liczba domen o długości , z racji własności operatora . Kolejną możliwą konwersją jest “doklejenie” wędrujących domen ’hh’ do powstających w dużych ilościach domen o długościach i . Zwiększa to prawdopodobieństwo powstania domen i (maksima na rys. 5.25b). Dokonuje się inwersja obsadzeń domen o długościach , , itd. na korzyść tych nieparzystych.
-
•
Równolegle operacje i “rozprowadzają” nierównowagowe domeny o nieparzystych długościach. W ten sposób równolegle do podziału domen dłuższych na krótsze następuje konwersja nadreprezentowanych najkrótszych domen w krysztale. Maksima z rys. 5.25b zanikają.
-
•
Następuje uformowanie ostatecznego rozkładu wielkości domen charakterystycznego dla kryształu całkowicie nieuporządkowanego.
a) 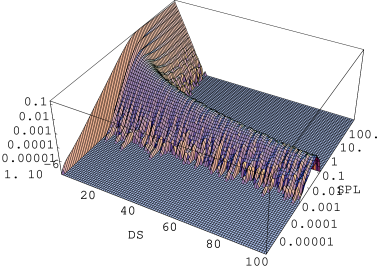

b) 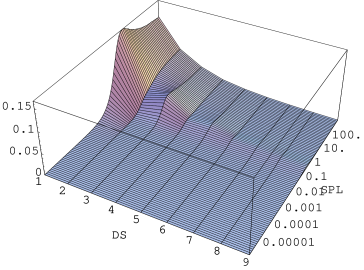
Figure 5.25: Rozkład długości domen (, pozioma oś liniowa) hexagonalnych w trakcie wprowadzania błędów ułożenia do krystalitu o strukturze i długości warstw. Zaawansowanie procesu tworzenia nieporządku wyrażone jest w przesunięciach na warstwę (Shifts Per Layer, , pozioma oś logarytmiczna), a jego kierunek zaznaczono strzałką. Liczba domen (oś pionowa) została unormowana do liczby domen w krystalicie. W skali logarytmicznej (a) widać fazę procesu (“kanał”), w której spektrum długości domen jest białe. W skali liniowej (b) widać maksima rozkładu dla domen o długościach nieparzystych (). W ostatniej fazie procesu () domeny budują wykładniczy rozkład długości - stan całkowitego nieuporządkowania.
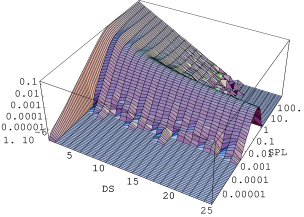
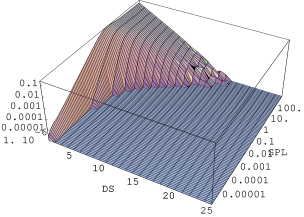
Proces wprowadzania błędów ułożenia do innych politypów (, , ) przebiega podobnie (rys. 5.26) z tym zastrzeżeniem, że bardzo długie domeny hexagonalne nie istnieją w nich od samego początku, więc przedmiotem dzielenia są raczej odpowiednie, długie domeny , i . Zachowują natomiast ważność uwagi na temat konwersji nadreprezentowanych najkrótszych domen hexagonalnych i tworzenia ostatecznego rozkładu domen struktury nieuporządkowanej. Proces rozporządkowywania struktury jest w ramach prezentowanego modelu całkowicie nieefektywny (rys. 5.26-3C). Można mówić o całkowitej rezystywności długich domen na wprowadzanie błędów ułożenia opisywaną metodą.
Równolegle do rozporządkowywania domen hexagonalnych następuje tworzenie (struktura ) lub przekształcenie (struktury , , ) rozkładu wielkości domen , rys. 5.26, kolumna II. W przypadku struktury każdy akt podziału domeny przy pomocy operatora tworzy dwie domeny o długości (warstwy typu ). Tak więc od samego początku procesu następuje przyrost ich liczby. W innych politypach domeny występują natywnie, np. w mają one długość , i następuje tylko ich konwersja do innych długości w okolicznościach podobnych do opisanych poprzednio dla domen hexagonalnych.
Pod koniec procesu rozporządkowywania () tworzy się końcowy rozkład wielkości domen odpowiadający strukturze całkowicie nieuporządkowanej, identyczny z rozkładem domen .
| Rozkład długości domen typu | Rozkład długości domen typu | |
|---|---|---|
 |
 |
|
 |
 |
|
 |
 |
5.5.6 Rozkład długości domen kryształu nieuporządkowanego
Rozkład długości domen całkowicie nieuporządkowanego kryształu jest rozkładem wykładniczym, co dobrze widać na rys. 5.26 (prosta linia rozkładu dla w skali logarytmicznej). Jest on dany wzorem:
| (5.10) |
gdzie . Lub podstawiając :
Jest to zgodne z przewidywaniami, ponieważ właśnie taki rozkład długości miałyby domeny budowane poprzez losowanie kolejnych warstw ( lub ) z równym prawdopodobieństwem1111 11 Prawdopodobieństwo, że wylosujemy warstwę : , że wylosujemy dwie pod rząd: , trzy: , itd. , czyli całkowicie nieuporządkowane.
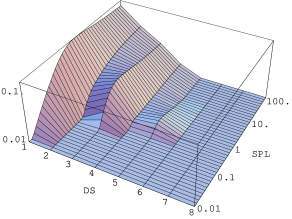
5.5.7 Rozkład długości domen w kryształach nanometrowych
Opis podany w 5.5.5 pozostaje w mocy także w odniesieniu do kryształów nanometrowych, z tym jednak zastrzeżeniem, że prawdopodobieństwo znalezienia domeny o długości większej niż rozmiar kryształu musi znikać. Nanokryształy są obiektami nie większymi niż kilkadziesiąt/kilkaset nanometrów, co odpowiada warstw. W takim przypadku, rys. 5.27, nie obserwujemy (poza największymi nanokryształami) fazy procesu rozporządkowania z bardzo szerokim rozkładem długości domen. W dalszym ciągu jest jednak obecna inwersja obsadzeń domen o długościach , , itd., na korzyść nieparzystych. Zostaje ona przełamana, tak jak w przypadku dużych kryształów, i tworzy się ostateczny, wykładniczy rozkład długości domen kryształu nieuporządkowanego.
a)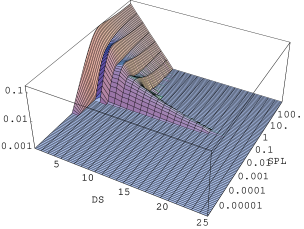
|
b)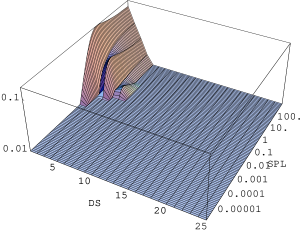
|
5.5.8 Rozkład długości domen a dyfrakcja
Postać rozkładu długości domen w warstwowym krysztale częściowo lub całkowicie nieuporządkowanym ma kapitalny wpływ na jego własności dyfrakcyjne. Na rys. 5.28a wykreślono profile trzech najsilniejszych refleksów (, i ) odpowiadające zmieniającym się rozkładom długości domen (5.28b) w trakcie symulowanego wprowadzania błędów ułożenia pod wpływem wysokiego ciśnienia niehydrostatycznego. Najsilniejszej degradacji w trakcie wprowadzania błędów ułożenia doznaje maksimum . Odpowiada ono rodzinie płaszczyzn przecinających warstwy hexagonalne pod kątem ostrym. Gładka w strukturze płaszczyzna jest “cięta” przez błędy ułożenia na coraz krótsze paski o szerokości kryształu i długości warstw, wynikającej z rozkładu długości domen (5.28b). Jest to wyraźnie widoczne w fazie wyrównywania inwersji obsadzeń domen o długościach , w której zmniejsza się liczba domen najdłuższych (). Podobny proces ma miejsce w odniesieniu do maksimum , odpowiadającego płaszczyznom prostopadłym do warstw hexagonalnych . Natężenie maksimum pozostaje w zasadzie niezmienione, jeśli nie liczyć silnie podniesionego poziomu tła. Tło jest bardzo charakterystycznym elementem profili dyfrakcyjnych struktur nieuporządkowanych a bierze się ze znacznej liczby obiektów zbyt krótkich (domeny o długościach 1 i 2) i ułożonych wzajemnie w sposób eliminujący konstruktywną interferencję. Końcowy stan całkowitego nieuporządkowania kryształ osiąga po wprowadzeniu ok. przesunięć na jedną warstwę. Oznaczałoby to przejście ponad stu dyslokacji w pojedynczym nanokrysztale, co jest liczbą dużą, ale jest możliwe (obserwuje się doświadczalnie, że dojście do stanu całkowitego nieuporządkowania wymaga przyłożenia ciśnienia rzędu kilkudziesięciu i/lub aktywacji termicznej).
Hipotezę o chwilowym istnieniu układu z przewagą domen o długościach nieparzystych wydają się potwierdzać jego szczególne własności dyfrakcyjne. Mianowicie, w stadium procesu rozporządkowania odpowiadającemu mamy (rys. 5.28b) już do czynienia z domenami całkowicie rozdrobnionymi, równie rozdrobnionymi jak dla - obie fazy procesu różnią się tylko proporcjami obsadzeń długości i . Odpowiadające tym fazom dyfraktogramy proszkowe (rys. 5.28a) różnią się jednak w sposób zasadniczy, co pozwala na ich odróżnienie, np. przez porównanie proporcji natężeń linii i . Zostało to zaobserwowane doświadczalnie.
 |
 |
| a) | b) |
5.5.9 Wyniki eksperymentalne
Dyfraktogramy proszkowe nanokryształów zostały zmierzone w komórce diamentowej we wzrastających ciśnieniach1212 12 Ciśnienie ustalano za pomocą pomiaru zmian wartości parametru sieci złota, którego drobiny zostały dodane do próbki azotku galu. Tak więc podane wartości odpowiadają ciśnieniu hydrostatycznemu działającemu na drobiny złota w ośrodku tworzonym przez nanokryształy ., aż do . Przedstawiono je w lewej kolumnie rys. 5.29. Jak widać na rys. 5.29, proporcje maksimów dyfrakcyjnych ulegają silnym zmianom: natężenia najsilniejszych linii i zmniejszają się, odpowiednio, do ok. i wyjściowych natężeń mierzonych względem linii . Linia nie zmienia się w trakcie procesu, jeśli nie liczyć podniesionego tła. Wszystkie linie dyfrakcyjne silnie poszerzają się. Zaobserwowane zmiany były trwałe: po zdjęciu ciśnienia kształt i proporcje natężeń pozostały takimi jak w . Pokazuje to, iż w materiale zaszły trwałe zmiany strukturalne, jakimi są wprowadzone w czasie procesu zagęszczania błędy ułożenia. W prawej kolumnie rys. 5.29 pokazano dyfraktogramy obliczone numerycznie ab initio tak, jak to opisano w paragrafie 4.2.1. Podczas obliczania dyfraktogramów użyto struktur krystalicznych , będących wynikiem wprowadzania błędów ułożenia do czystej struktury za pomocą przesunięć pojedynczych warstw tak, jak to opisano w paragrafie 5.5.3.
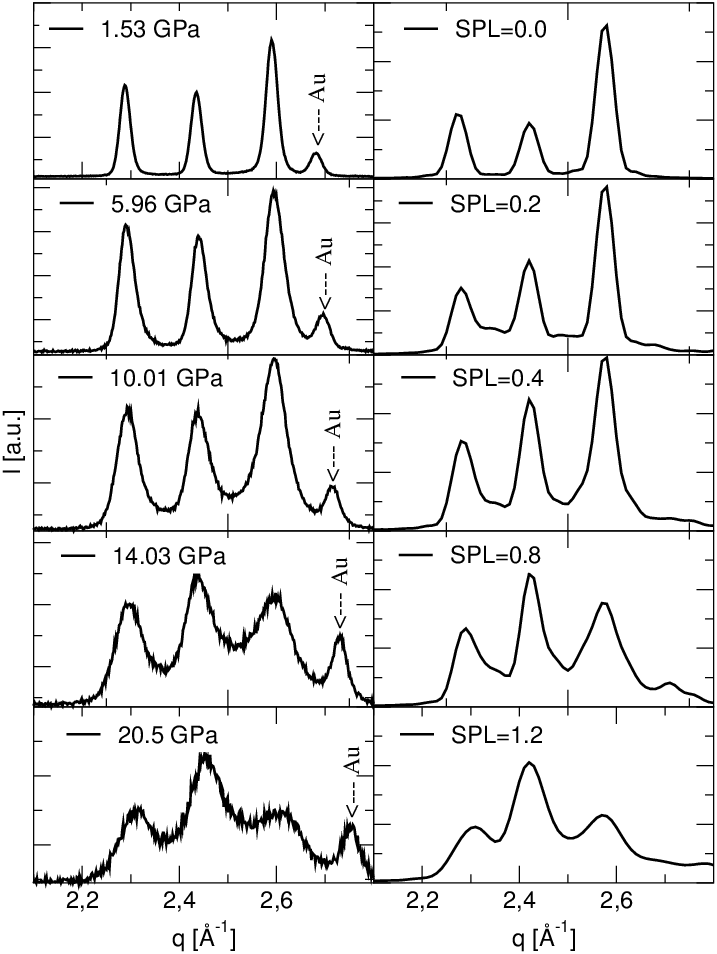 |
Porównując pary dyfraktogramów w kolejnych wierszach rys. 5.29 widać, że obliczenia ab initio (krzywe po prawej) odtwarzają jakościowo przebieg krzywych doświadczalnych (po lewej). Wraz ze wzrastającym ciśnieniem refleksy widoczne na dyfraktogramach doświadczalnych poszerzają się i podnosi się natężenie tła. Odpowiednie krzywe teoretyczne zachowują się analogicznie. Różnice natężeń refleksów widoczne przy niskich ciśnieniach wynikają z przyjętego kształtu i rozmiaru ziaren. W symulacji używano kulistych modeli krystalitów, w których mamy tę samą liczbę atomów wzdłuż każdego kierunku krystalograficznego. Rzeczywiste nanokryształy rosną zazwyczaj w postaci kolumn hexagonalnych, co drastycznie zmniejsza ilość par atomowych leżących w kierunkach ukośnych, również w kierunku , zmniejszając tym samym natężenie odpowiedniego refleksu. Różnica szerokości linii jest związana z wielkością modeli użytych podczas obliczeń dyfrakcji, którą ustalono na Å z powodów ograniczeń numerycznych. Rozmiar mierzonych nanokryształów jest większy, stąd szerokość linii dyfrakcyjnych - mniejsza. Wraz ze wzrostem ciśnienia (kolejne wiersze rys. 5.29) wpływ opisanych różnic kształtu i rozmiaru ziarna na obraz dyfrakcyjny zmniejsza się. Po (lewej) stronie rzeczywistych nanokryształów razem z ciśnieniem pojawiają się naprężenia sieci krystalicznej nieco poszerzając linie dyfrakcyjne (przejście od do ). Po (prawej) stronie krzywych symulowanych struktura błędów ułożenia (rozkład długości domen ) staje się najsilniejszym czynnikiem kształtującym obraz dyfrakcyjny począwszy od przejścia z do (co odpowiada zmniejszeniu hexagonalności z do ). Od tego miejsca (, , ) wpływ kształtu i rozmiaru ziaren staje się tylko poprawką do kształtu dyfraktogramu określanego przede wszystkim przez strukturę błędów ułożenia gdyż koherentnie rozpraszające domeny stają się małe (por. rys. 5.28b) w porównaniu do rozmiaru nanokryształu. Nie można również wykluczyć pękania rozciągłych w przestrzeni płytek i igieł w wysokich ciśnieniach niehydrostatycznych, co także zbliża proporcje maksimów mierzonych do symulowanych. W ciśnieniu nanokryształy osiągają stan bliski całkowitemu nieuporządkowaniu (hexagonalność ) zaś rozkład długości domen staje się wykładniczy (por. paragrafy 5.5.6 i 5.5.7).
5.5.10 Wnioski
Zaproponowany prosty model poprzez przesunięć pojedynczych warstw jest - jak sądzimy - jednym z prawdopodobnych mechanizmów relaksacji naprężeń w strukturach . Przemawiają za tym dwa fakty: najmniejsza energia przejścia dyslokacji właśnie wzdłuż płaszczyzn oraz obecność w doświadczalnych danych dyfrakcyjnych charakterystycznych zmian powodowanych przez błędy ułożenia, tworzące się na skutek tych dyslokacji. Na przykładzie nanokryształów pokazano, że obserwowane w wysokich ciśnieniach gwałtowne zmiany własności dyfrakcyjnych materiału można wyjaśnić za pomocą prezentowanego modelu. Mając pełną świadomość, że model ten nie uwzględnia szeregu zjawisk, które z pewnością zachodzą równolegle do wyżej opisanych (jak np. ścinanie kryształów wzdłuż warstw ), można powiedzieć, że przesunięcia pojedynczych warstw są jednym z mechanizmów relaksacji naprężeń w nanokryształach o strukturze i pokrewnych (np. strukturach najgęstszego upakowania z błędami ułożenia, jak czy ). Pokazano, że opisywane w pracy (§4.2) metody obliczeń ab initio w dyfrakcji proszkowej pozwalają śledzić przemiany dowolnie złożonych struktur atomowych, w tym ewolucję jednowymiarowego nieuporządkowania nanokryształów, niemożliwe do uchwycenia tradycyjnymi technikami.